Clockophagy,一種有利于鐵死亡的新型選擇性自噬過程
鐵死亡是一種由鐵依賴性脂質過氧化驅動的非凋亡調節性細胞死亡。自噬涉及溶酶體降解途徑,可以促進或阻止細胞死亡。高水平的自噬與鐵死亡有關,但這種關系背后的機制還沒有明確的解釋。而文章“Clockophagy is a novel selective autophagy process favoring ferroptosis”正是針對此所做的研究,研究揭示了一條由ARNTL的自噬去除開始,最終促進鐵死亡誘導新的途徑。

摘要:
文章的作者利用人類癌細胞株和小鼠腫瘤模型中研究了自噬對鐵死亡的作用。研究發現,自噬作用對核心晝夜節律蛋白ARNTL的選擇性降解,即“鐘表性吞噬”(clockophagy),對鐵死亡起關鍵作用。SQSTM1是負責ARNTL自噬降解的貨物受體。ARNTL通過抑制EGLN2的轉錄來抑制鐵死亡,從而激活促生存轉錄因子HIF1A。阻止ARNTL降解或抑制EGLN2激活的遺傳或藥物干預減少,而不穩定的HIF1a增加,鐵死亡腫瘤細胞死亡。
一、鐵死亡中ARNTL的選擇性降解
1型鐵死亡激活劑:系統XC?抑制劑,如Erastin,柳氮磺胺吡啶和索拉非尼。
2型鐵死亡激活劑:是Gpx4抑制劑,包括RSL3和FIN56。
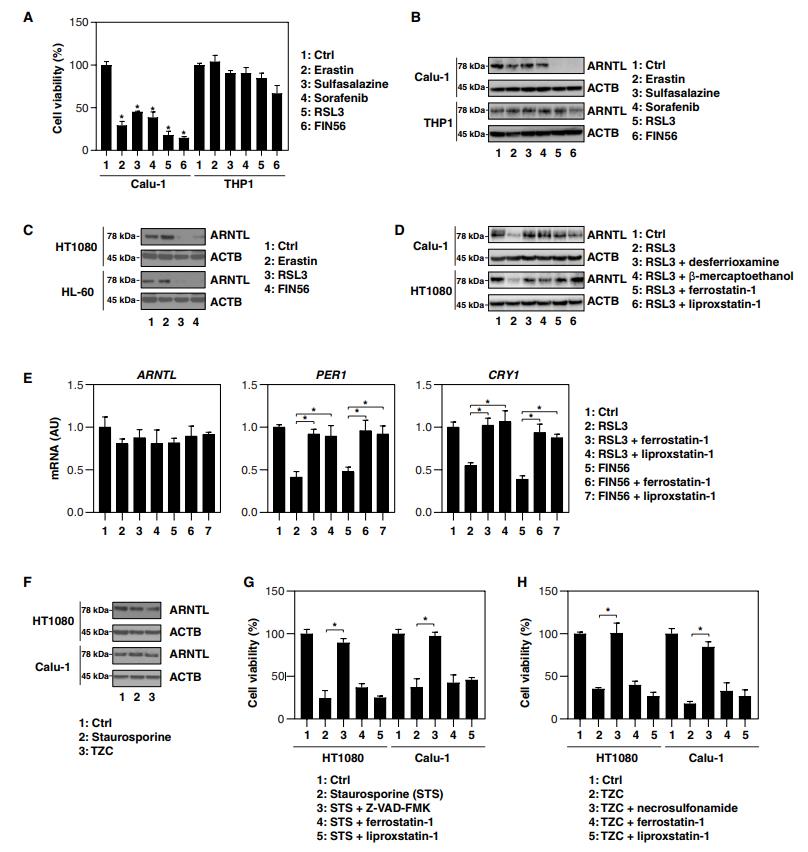
Calu-1(一種人類非小細胞肺癌細胞系)細胞對鐵死亡激活劑敏感,而THP1(一種人類急性單核細胞白血病細胞系)對它們具有抗性(圖1A)。免疫印跡分析顯示,在Calu-1細胞中,ARNTL的蛋白表達被2型激活劑抑制,而1型激活劑不行(圖1B)。2型激活劑也抑制了其他鐵死亡敏感細胞系——HT1080(人纖維肉瘤細胞系)和HL-60(人類早幼粒細胞白血病細胞系)中ARNTL蛋白的表達(圖1C)。鐵死亡抑制劑(去鐵胺,β-巰基乙醇,ferrostatin-1和liproxstatin-1),在Calu-1和HT1080細胞中逆轉了RSL3誘導的ARNYL蛋白下調(圖1D)。以ferrostatin-1和Liproxstatin-1為變量, RSL3和FIN56誘導沒有顯著改變ARNTL的mRNA水平(圖1E)。相反,RSL3和FIN56下調ARNTL靶向生物鐘基因如PER1和CRY1的mRNA改變(圖1E)。凋亡誘導劑(staurosporine)或壞死性凋亡誘導劑 TCZ[TNF(腫瘤壞死因子),Z-VADFMK和CHX]不能誘導ARNTL降解(圖1F)。而Z-VAD-FMK(一種泛半胱氨酸酶抑制劑)和necrosulfonamid[一種針對MLKL(混合譜系激酶結構域樣偽激酶)的壞死型凋亡抑制劑],則分別抑制staurosporine和TZC誘導的細胞死亡(圖1G,H)。
這些發現表明2型鐵死亡激活劑選擇性地誘導ARNTL蛋白降解。
二、SQSTM1是自噬ARNTL降解的受體
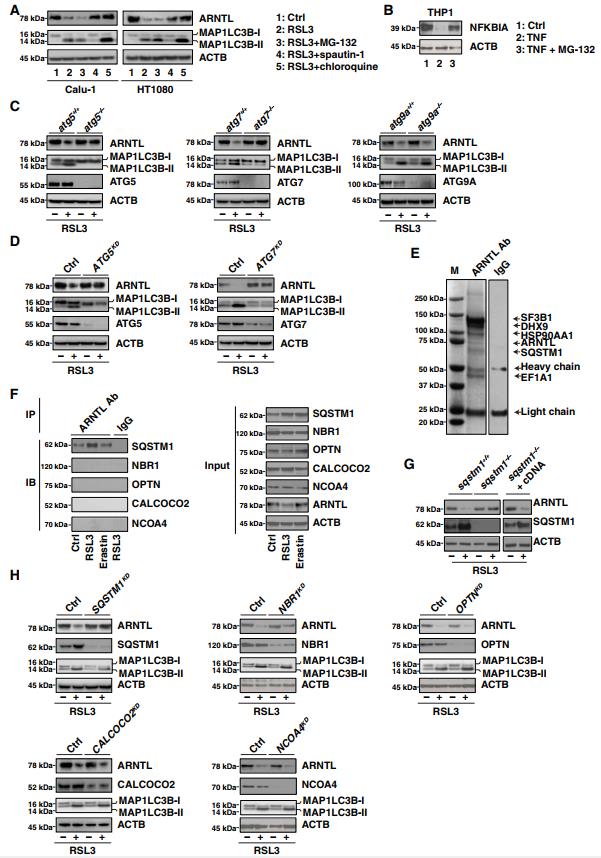
哺乳動物細胞有兩種細胞內蛋白降解途徑,即泛素-蛋白酶體系統和自噬。蛋白酶體抑制劑Mg-132未能阻斷RSL3誘導的CALU-1和HT1080細胞中ARNTL蛋白的降解(圖2A)。作為陽性對照,MG-132抑制TNF誘導的NFKBIA/IκBα(核因子?B抑制劑?)在THP1細胞中的降解(圖2B),這與以前的發現一致,即TNF誘導的NFKBIA降解是蛋白酶體依賴性的。與MG-132不同,spautin-1(早期自噬抑制劑)和氯喹(晚期自噬抑制劑)可保護Calu-1和HT1080細胞免受RSL3誘導的ARNTL蛋白降解(圖2A)。這些發現表明自噬,而不是蛋白酶體,可能有助于ARNTL蛋白在鐵死亡過程中的降解。
我們接下來討論了哪個自噬途徑參與了ARNTL降解的調節。ATG5和ATG7是饑餓誘導的自噬小體形成所必需的。ATG5或ATG7的敲除抑制了MAP1LC3B(微管相關蛋白1輕鏈3β)-I向MAP1LC3B-II(自噬小體形成的標志物)的轉化,以及對RSL3作出反應的小鼠胚胎成纖維細胞(MEFs)中ARNTL的降解(圖2C)。同樣,特異性短發夾RNA(ShRNAs)對ATG5或ATG7的敲除抑制了HT1080細胞中RSL3誘導的MAP1LC3B-II產生和ARNTL降解(圖2D)。然而,跨膜核心ATG蛋白Atg9a的敲除未能阻止這些過程(圖2C),而ATG蛋白從囊泡向自噬體提供膜。因此,ATG5和ATG7,而不是ATG9A,有助于自噬小體的形成和隨后在RSL3誘導的鐵死亡過程中ARNTL的自噬降解。特定的貨物受體參與選擇性自噬。SQSTM1是一種多功能的貨物受體,參與泛素化底物的自噬降解,包括蛋白質和細胞器。質譜分析確定SQSTM1在正常條件下是ARNTL的相互作用物(圖2E)。免疫沉淀分析表明,在RSL3誘導的而不是Erastin誘導的鐵死亡中,SQSTM1-ARNTL相互作用增加(圖2F)。相反,在沒有或存在RSL3的情況下,ARNTL未能與其他貨物受體如NBR1(NBR1,自噬貨物受體),OPTN(Optineurin),CALCOCO2/NDP52(鈣結合和卷曲螺旋結構域2)和NCOA4(核受體共激活劑4) 結合。Sqstm1缺失減少了MEF中RSL3誘導的ARNTL降解(圖2G)。相反,SQSTM1?/?MEFs中Sqstm1互補DNA的表達恢復了RSL3誘導的ARNTL降解(圖2G)。shRNA對Sqstm1(而不是Nbr1,Optn,Ccolco2或Ncoa4)的敲除也阻止了HT1080細胞中RSL3誘導的ARNTL蛋白的降解(而不是MAP1LC3BII表達)(圖2H)。免疫共沉淀分析進一步發現,在沒有或存在RSL3的情況下,SQSTM1的泛素相關(UBA)結構域是SQSTM1-ARNTL相互作用所必需的(圖S1)。與饑餓誘導的大量自噬中發生的SQSTM1的下調不同,在RSL3誘導的選擇性自噬中觀察到SQSTM1的上調(圖2,F到H),這表明SQSTM1的變化可能是細胞類型和環境特異性的。
共聚焦顯微鏡分析進一步發現,RSL3增強了MAP1LC3B、SQSTM1、LAMP2(溶酶體相關膜蛋白2)和ARNTL之間的共定位,而Erastin不能(圖S2A到C)。此外,氯喹增強RSL3誘導的ARNTL,MAP1LC3B和SQSTM1(而不是LAMP2)之間的共定位(圖S2A到C)。溶酶體組分的Western blot分析證實RSL3可導致ARNTL的表達增加,而Erastin不能(圖S2D)。總之,這些發現表明SQSTM1是RSL3誘導的鐵死亡過程中溶酶體中ARNTL自噬降解的直接受體。
三、自噬介導的ARNTL降解促進鐵死亡
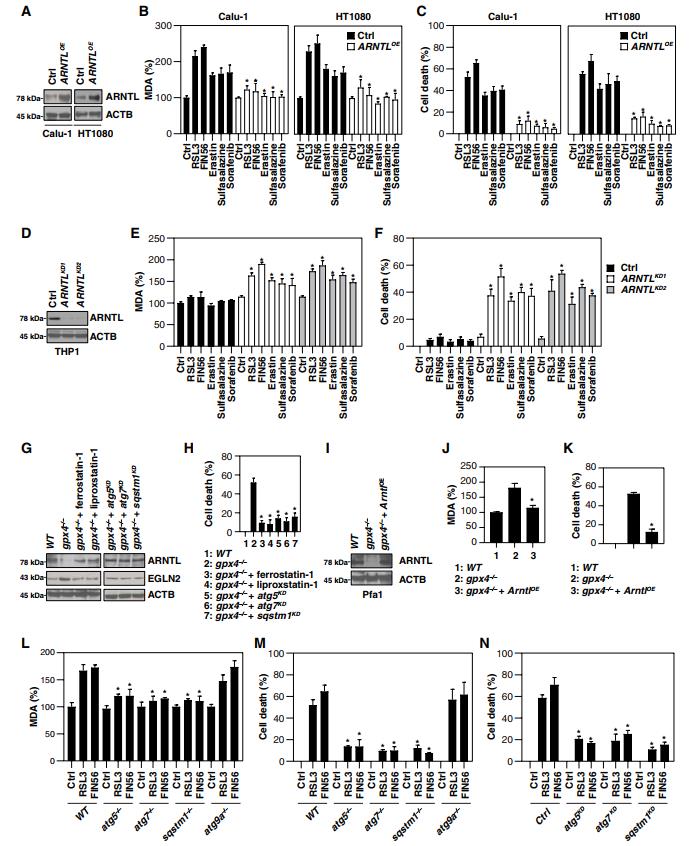
我們接下來研究了自噬介導的ARNTL降解對鐵死亡的影響。首先,ARNTL通過基因轉染在鐵死亡敏感細胞系(Calu-1和HT1080)中過度表達(圖3A)。ARNTL的過度表達減少了RSL3-和FIN56誘導的丙二醛(MDA;脂質過氧化的最終產物)的產生(圖3B)和細胞死亡(圖3C)。雖然ARNTL的基本表達不受1型激活劑(erastin,柳氮磺胺吡啶和索拉非尼)的影響(圖1B),但ARNTL的過度表達抑制了Calu-1和HT1080細胞中1型激活劑誘導的細胞死亡和MDA產生(圖3,B和C),表明ARNTL表達閾值對1型激活劑誘導的鐵死亡有影響。相反,由兩個不同的shRNAs(圖3d)敲除ARNTL恢復了THP1細胞中由1型和2型鐵死亡激活劑誘導的的MDA產生(圖3e)和細胞死亡(圖3f),表明ARNTL的耗盡可以克服對鐵死亡的抗性。
Gpx4是各種應激條件下脂質過氧化的中樞負調節因子。先前的研究表明,Gpx4的誘導性敲除導致Pfa1細胞中的鐵死亡細胞死亡。類似于RSL3處理(圖2A),Gpx4的敲除增加了MAP1LC3B-II的產量(圖S3)。我們觀察到gpx4?/? Pfa1細胞中ARNTL的下調(圖3G)和細胞死亡(圖3H)被鐵死亡抑制劑(例如,ferrostatin-1和Liproxstatin-1)或Atg5,Atg7或Sqstm1的敲除逆轉。轉染強制過表達ARNTL(圖3i)降低了gpx4?/?Pfa1細胞中MDA產生(圖3j)和細胞死亡(圖3k),表明Gpx4耗盡介導的arnt蛋白降解是鐵死亡所必需的。此外,Atg5、Atg7或Sqstm1(而不是Atg9a)的敲除減少了MEF中RSL3和FIN56誘導的MDA產生(圖3L)和的細胞死亡(圖3M)。Atg5、Atg7或Sqstm1的敲除也減少了RSL3和FIN56誘導的HT1080細胞中的細胞死亡(圖3N)。總之,這些發現表明自噬介導的ARNTL降解通過激活脂質過氧化來促進鐵死亡。
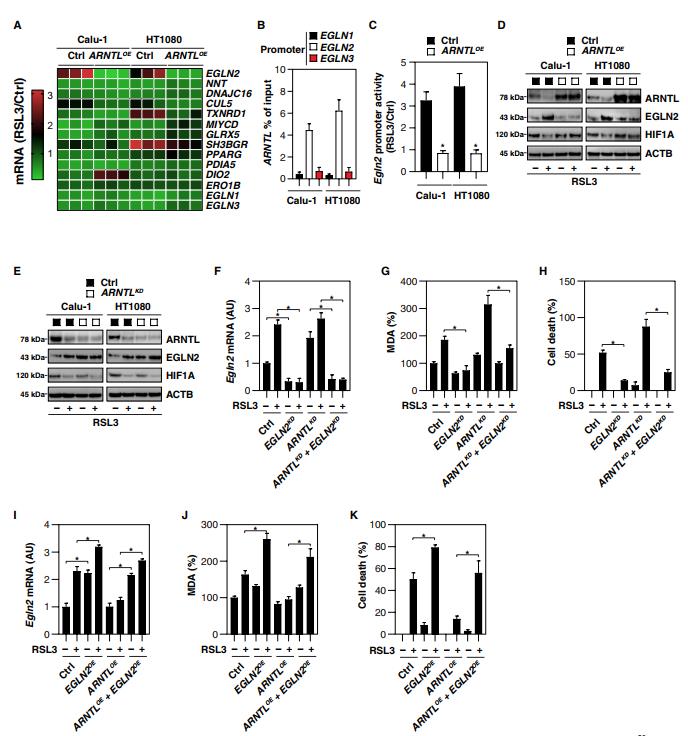
四、ARNTL介導的EGLN2下調阻斷鐵死亡
ARNTL是一種晝夜節律轉錄因子,主要通過在其啟動子中結合E-box基序(CAGCTG或CACGTG)來調節基因表達。真核生物啟動子數據庫(https://epd.vital-it.ch/index.php)列出了1666個帶有E-box基序的人類基因。DAVID(注釋、可視化和集成發現數據庫;在線工具對基因簇的基因本體分析進一步顯示12個含有E-box的基因——EGLN2,NNT,DNAJC16 ,CUL5,TXNRD1,MLYCD,GLRX5,SH3BGR,PPARG,PDIA5,DIO2and ERO1B——可能參與氧化應激的調節。我們使用定量聚合酶鏈反應(qPCR)來確定這些基因是否作為鐵死亡的一部分由ARNTL直接控制。EGLN2 mRNA在對RSL3作出反應的Calu-1和HT1080細胞中均上調(圖4A)。相反,ARNTL的過度表達阻斷了在Calu-1和HT1080細胞中RSL3誘導的EGLN2上調(圖4A)。編碼其他EGLN家族成員,包括EGLN1和EGLN3的mRNAs在用RSL3處理和/或ARNTL過度表達后,其豐度沒有經歷任何重大變化(圖4A)。因此,染色質免疫沉淀(ChIP)試驗還發現,在Calu-1和HT1080細胞中,ARNTL結合到EGLN2的啟動子(而不是EGLN1和EGLN3)上(圖4B)。報告基因(Fig,4C)和Western blot(Fig,4D)分析確定,在RSL3誘導的鐵死亡中,EGLN2被ARNTL抑制。因此,shRNA對ARNTL的敲除增加了Calu-1和HT1080細胞中EGLN2的表達(圖4E)。除了 ARNTL下調(圖3G),gpx4?/? Pfa1細胞中的EGLN2 上調調控(圖3G)也被鐵死亡抑制劑(例如, ferrostatin-1 和Liproxstatin-1)或Atg5,Atg7或Sqstm1的敲除逆轉,支持自噬調節ARNTL和EGLN2表達以響應Gpx4耗盡誘導的脂質過氧化。
為了確定EGLN2的上調是否有助于鐵死亡,我們在Calu-1和HT1080細胞中通過shRNA敲除了EGLN2。EGLN2表達的抑制(圖4F)限制了對照和ARNTL敲除HT1080細胞中RSL3誘導的MDA產生(圖4G)和細胞死亡(圖4H)。相反,轉染強制EGLN2過度表達(圖4I)在對照和過度表達ARNTL的HT1080細胞中都增加了RSL3誘導的MDA產生(圖4J)和細胞死亡(圖4K)。總之,這些發現支持ARNTL通過下調EGLN2表達抑制鐵死亡的假設。
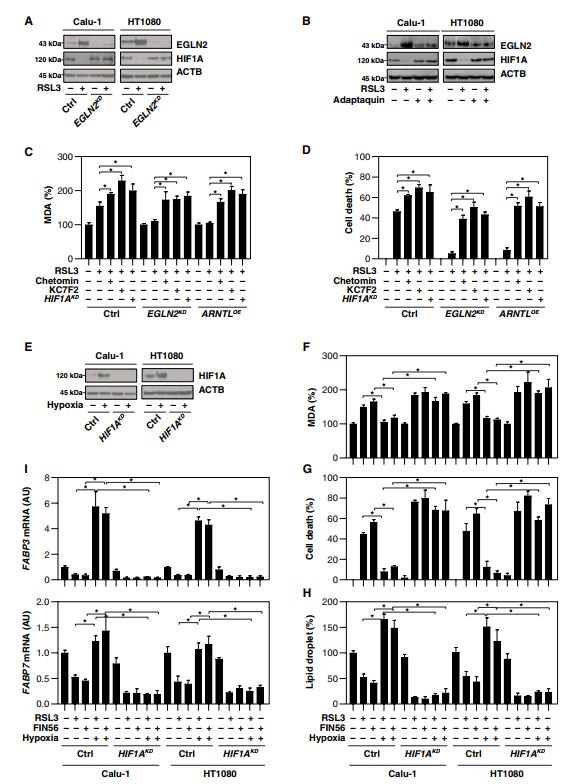
五、EGLN2介導的HIF1A下調促進鐵死亡
HIF1A(缺氧誘導因子1亞基α)是一種轉錄因子,調節微環境中氧可利用性降低的穩態反應。鑒于EGLN2的主要功能是抑制HIF1a的激活,我們試圖確定HIF1a的表達是否受ARNTL介導的EGLN2下調調控。ARNTL過表達抑制EGLN2的表達,而EGLN2又在RSL3處理后的Calu-1和HT1080細胞中維持HIF1a的表達(圖4D)。相反,ARNTL敲除促進EGLN2表達,與RSL3處理后Calu-1和HT1080細胞中HIF1A表達降低相關(圖4E)。ARNTL的敲除在基線條件下部分降低了HIF1a表達(圖4E),表明其他非ARNTL途徑可能有助于HIF1a基本表達。通過特異性shRNA(圖5A)或應用adaptaquin(圖5B)對EGLN的遺傳或藥理作用抑制,增加了RSL3處理的Calu-1和HT1080細胞中HIF1A的表達。總之,這些發現表明ARNTL通過下調EGLN2在RSL3誘導的鐵死亡中促進HIF1aA的表達。
HIF1a降解是由泛素-蛋白酶體途徑介導的。正如預期的那樣,MG-132抑制RSL3誘導的HT1080細胞中HIF1a下調,但不能抑制ARNTL下調和EGLN2上調(圖S4A)。HIF1A也可以被鐵螯合劑去鐵胺誘導。正如預期的那樣,去鐵胺在RSL3處理后恢復了ARNTL和HIF1A蛋白水平,降低了HT1080細胞中EGLN2的表達(圖S4B)。與RSL3處理不同(圖S4A和B),erastin處理不改變EGLN2和HIF1A的表達(圖S4C)。
我們接下來試圖研究HIF1A在鐵死亡中的作用。HIF1A抑制劑(例如chetomin和KC7F2)的應用或HIF1A敲除在EGLN2敲除或ARNTL過表達的HT1080細胞中恢復了RSL3誘導的MDA產生(圖5C)和細胞死亡(圖5D)。此外,低氧預處理誘導HIF1A表達(圖5E),因為它限制了RSL3-和FIN56誘導的Calu-1和HT1080細胞的MDA產生(圖5F)和細胞死亡(圖5G)。相反,通過缺氧誘導HIF1A激活以響應RSL3和FIN56(圖5H),脂滴的形成--中性脂質儲存的細胞內部位--得以恢復。此外,負責脂肪酸攝取和脂質儲存的兩個關鍵HIF1a靶基因FABP3和FABP7的mRNA表達通過在RSL3和FIN56處理后在的Calu-1和HT1080細胞中的HIF1A激活而恢復(圖5I)。總之,這些發現證實HIF1A是鐵死亡中的促生存因子,而EGLN2介導的HIF1a下調促進鐵死亡。
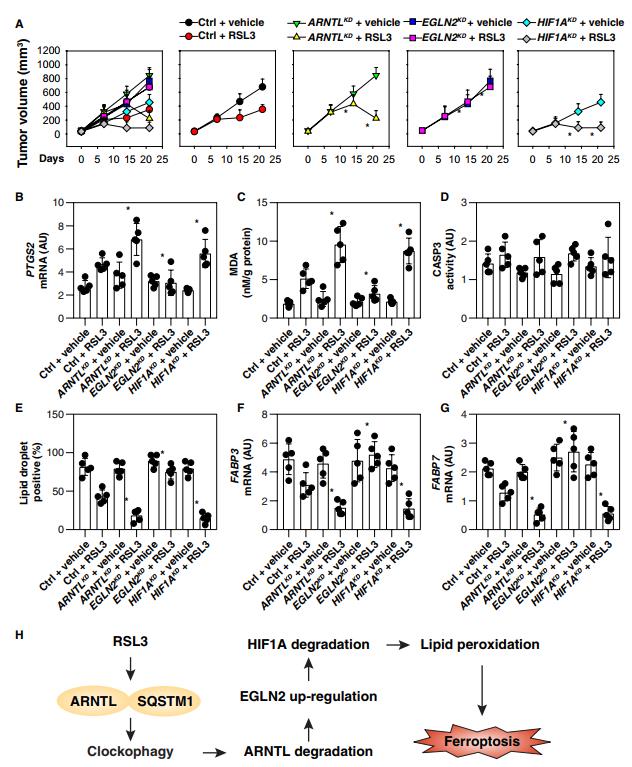
六、ARNTL途徑在體內調節鐵死亡
為了確定ARNTL途徑是否也調節體內腫瘤對鐵死亡激活劑的敏感性,我們皮下接種ARNTL,EGLN2和HIF1A敲除的HT1080細胞到免疫缺陷小鼠的右側。從第7天開始,這些小鼠被系統地用(1S,3R)-RSL3,一種代謝穩定的RSL3衍生物,適合體內實驗,治療2周。與對照shRNA組相比,RSL3治療有效地減小了攜帶ARNTL或HIF1A敲除細胞的小鼠形成的腫瘤的大小(圖6A)。相反,攜帶EGLN2基因敲除細胞的小鼠對RSL3治療有抵抗力。PTGS2(前列腺素內過氧化物合成酶2)表達的qPCR分析表明,ARNTL和HIF1A敲除增加而EGLN2敲除抑制體內鐵死亡。PTGS2是體內鐵死亡評估(圖6B)和MDA定量的標志(圖6C)。相反,在這些基因缺失的腫瘤中,CASP3 (caspase 3;凋亡標記)活性(凋亡標記)沒有被RSL3改變(圖6D)。值得注意的是,在RSL3治療后,ARNTL和HIF1A敲除HT1080腫瘤中脂肪滴的形成(圖6e)及FABP3(圖6f)和FABP7(圖6g)的mRNA表達降低。相反,它們在EGLN2敲除HT1080細胞中基本上沒有受到影響(圖6,E到G)。
Spautin-1(自噬抑制劑),adaptaquin(EGLN2抑制劑)和Liproxstatin-1(鐵死亡抑制劑)阻斷RSL3介導的腫瘤生長抑制(圖S5A),一種與PTGS2 mRNA表達降低相關的效應(圖S5B),和MDA產生(圖S5C)。相反,HIF1A抑制劑chetomin增強了RSL3的抗癌活性(圖S5A),Ptgs2 mRNA表達(圖S5B)和MDA產生(圖S5C),而不影響CASP3活性(圖S5D)。同時,脂滴形成(圖2S5E)、FABP3(圖5S5F)和FABP7(圖S5G)mRNAs的豐度被chetomin降低,與觀察到的這些參數響應于spautin-1、adaptaquin和liproxstatin-1而增加的情況形成對比。總之,這些發現表明ARNTL在體內拮抗RSL3介導的鐵死亡的抗癌活性。