






SNHG7促進腫瘤耐藥和巨噬細胞M2極化
小核仁RNA宿主基因7 (small nucleolar RNA host gene 7,SNHG7) 的衍生lncRNA在包括肺腺癌(LUAD)在內的多種癌癥中已被廣泛報道。然而,lncRNA SNHG7是否參與LUAD的多西他賽耐藥尚不清楚。本文發現外泌體介導SNHG7的轉移增強了肺腺癌的多西他賽耐藥。本文于2021年10月發表在《Cancer Letter》IF:8.679雜志上。
技術路線:

主要實驗結果:
首先檢測SNHG7是否和LUAD細胞抵抗多西他賽有關,結果顯示與相應的親代細胞相比,SNHG7在多西他賽耐藥的LUAD細胞系(H1299/DTX and SPC-A1/DTX)中高表達(圖1A)。敲除SNHG7后,H1299/DTX and SPC-A1/DTX對多西他賽的耐藥性顯著降低(圖1B-C),表明SNHG7增強LUAD細胞對多西他賽的抗性。此外,SNHG7在耐藥細胞和相應的親代細胞中都主要分布于細胞質中,表明其增強耐藥性是通過轉錄后調控。

圖1 SNHG7增強LUAD細胞對多西他賽的抗性
2、SNHG7增強LUAD細胞的多西他賽抗性,誘導巨噬細胞M2極化
為了研究SNHG7是否影響多西他賽對LUAD細胞的生物學功能,在有無多西他賽處理下進行功能實驗。如圖2A-C所示,陳曼SNHG7降低了0或100μg/L多西他賽暴露后的H1299/DTX and SPC-A1/DTX細胞的增殖,但增加了細胞凋亡。在小鼠體內,多西他賽抑制腫瘤生長,減小腫瘤體積和重量,而這種作用在SNHG7敲除后進一步被放大(圖2D-F)。因此,SNHG對多西他賽在LUAD細胞中的抗腫瘤作用有負面影響。
眾所周知M2極化的腫瘤相關巨噬細胞會促進腫瘤進展和化學抗性。所以作者想知道SNHG7是否會影響巨噬細胞M2極化。結果顯示IL-3處理巨噬細胞增加了M2標志物的表達,包括CD206和ARG1,但是敲除SNHG7會反轉這種趨勢(圖2G-H)。此外,IL-3處理增加了CD11b+CD206+標記的巨噬細胞比例,但敲除SNHG7會反轉這種作用(圖2I)。這些結果表明SNHG7正向調節巨噬細胞M2極化。
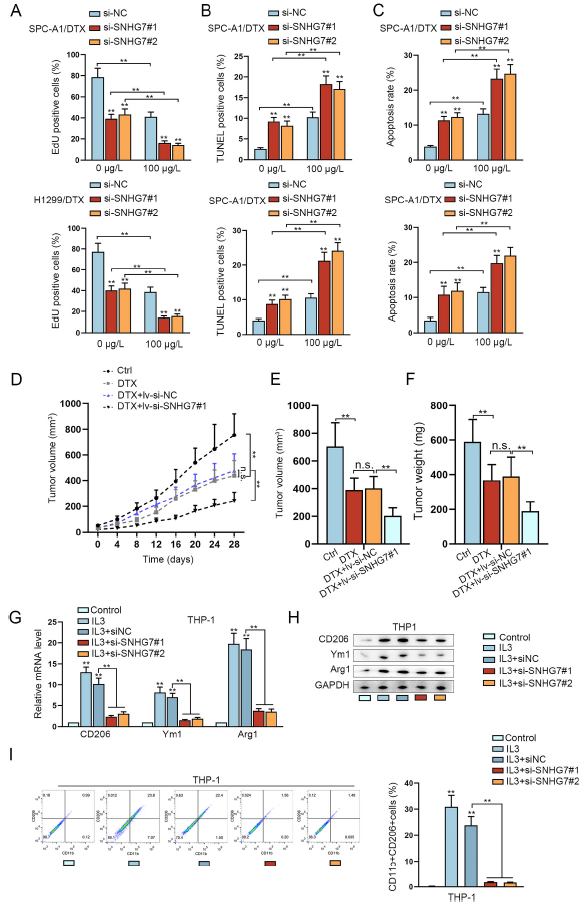
圖2 SNHG7增強LUAD細胞的多西他賽抗性,誘導巨噬細胞M2極化
3、SNHG7通過調節ATG5和ATG12誘導多西他賽耐藥的LUAD細胞自噬
自噬在調控腫瘤細胞耐藥中發揮著重要作用,所以作者猜測SNHG7可能通過自噬促進耐藥,于是檢測了敲除SNHG7后耐藥細胞H1299/DTX和SPC-A1/DTX中自噬相關基因的表達。如圖3A所示,WB表明敲除SNHG后暴露0或100μg/L多西他賽 H1299/DTX and SPC-A1/DTX細胞中LC3I/II, ATG5, ATG12的表達顯著降低,但不影響ATG7和ULK1;100μg/L多西他賽處理顯著降低了H1299/DTX and SPC-A1/DTX細胞中LC3-II/LC3-1, ATG5, ATG7, ATG12, and ULK1水平,但同時敲除SNHG7只增強了多西他賽導致的LC3I/II, ATG5, ATG12的表達降低作用。這表明SNHG7正調節自噬通過ATG5和ATG12。此外,免疫熒光顯示敲除SNHG7可以減少自噬小體和ATG5和ATG12蛋白水平,但是沒有改變其mRNA水平,并且熒光素酶實驗顯示SNHG7不能改變ATG5和ATG12的啟動子熒光素酶活性(圖3B-D),表明SNHG7不是在轉錄水平調控ATG5和ATG12。
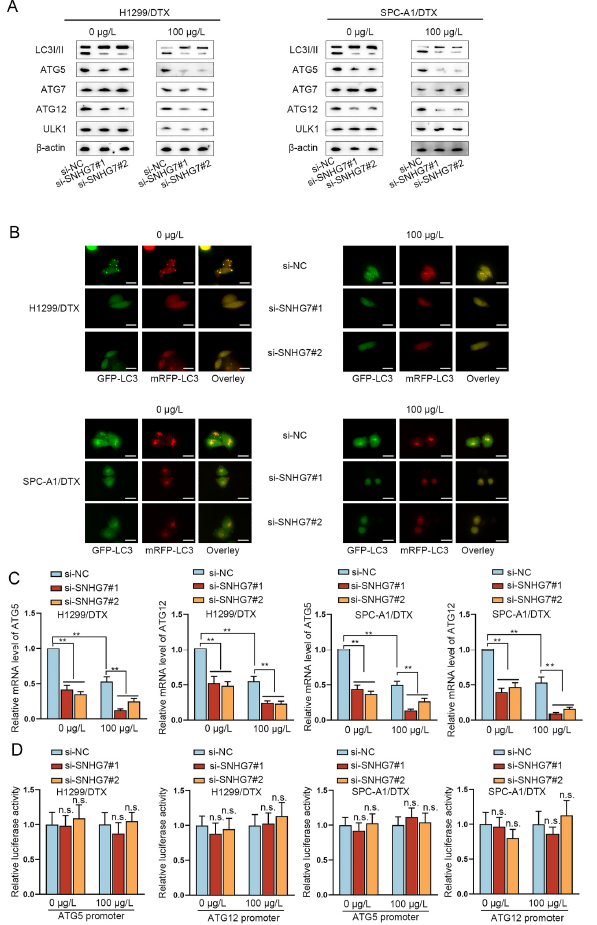
圖3 SNHG7通過調節ATG5和ATG12誘導多西他賽耐藥LUAD細胞自噬
4、SNHG7招募HuR以穩定ATG5和ATG12從而影響LUAD細胞的增殖、凋亡和多西他賽耐藥
接下來,研究SNHG7如何調控ATG5和ATG12。首先進行RNA pulldown實驗獲得與antisense比較在,senseSNHG7中差異的蛋白條帶,然后用MS識別差異條帶,獲得與SNHG7相互作用的蛋白,結果發現CUL4A和HuR在senseSNHG7中顯著富集(圖4A-B)。RIP證實SNHG7可以被HuR抗體富集沉淀(圖4B-C)。RIP證明HuR和ATG5和ATG12有結合作用(圖4D-E),并且,敲除SNHG7減少了ATG5和ATG12和HuR的結合(圖4F)。此外,敲除SNHG7或HuR的表達都會顯著降低ATG5和ATG12的mRNA穩定性(圖4G-H)。以上結果表明SNHG7招募HuR以穩定ATG5和ATG12。
之后作者通過拯救實驗證明SNHG是通過 ATG5和ATG12影響LUAD細胞的增殖、凋亡和多西他賽耐藥性的(結果未展示)。

圖4 SNHG7招募HuR穩定ATG5和ATG12
5、多西他賽耐藥LUAD細胞分泌的外泌體SNHG促進LUAD的耐藥性和巨噬細胞M2極化
如圖5A-D所示,分離獲得了耐藥性LUAD細胞系H1299/DTX and SPC-A1/DTX的外泌體,隨后這些被PKH67標記的外泌體顯示分布在H1299/DTX and SPC-A1/DTX的細胞質中。之后,將這些外泌體和SNHG7敲除或未敲除的巨噬細胞共孵育。結果發現外泌體處理組M2標志物顯著上調,但是當同時敲除SNHG7后,這種上調作用被減弱(圖5E-F)。類似的,CD11b+CD206+巨噬細胞比例在外泌體處理組顯著上調,在SNHG7+外泌體組顯著回落(圖5G)。表明耐藥性LUAD細胞來源外泌體SNHG7促進LUAD細胞的耐藥性和巨噬細胞M2極化。

圖5來自多西他賽耐藥LUAD細胞的外泌體SNHG7促進LUAD細胞的多西他賽耐藥和巨噬細胞M2極化
6、SNHG7通過招募CUL4A增加PTEN泛素化水平,激活PI3K/AKT通路介導巨噬細胞M2極化
隨后探究SNHG7調控M2極化的機制。作者首先篩選了9條常見的信號通路,并進行熒光素酶實驗,發現只有PI3K/AKT信號通路的熒光素酶信號在SNHG敲除后顯著下降(圖6A)。隨后WB驗證表明敲除SNHG7會顯著抑制外泌體處理的巨噬細胞AKT的磷酸化,但對其mRNA的表達無影響(圖6B-C)。亞細胞定位也顯示外泌體處理的巨噬細胞中SNHG7主要定位與細胞質(圖6D)。之后過表達SNHG7的同時用蛋白合成抑制劑CHX處理外泌體孵育后的巨噬細胞,發現過表達SNHG7后PTEN蛋白穩定性下降,并且外泌體處理的巨噬細胞中PTEN的泛素化增加了,而MG132阻斷了這種SNHG過表達對PTEN泛素化的抑制作用(圖6E)。
此外,之前的MS鑒定到的CUL4A是一個泛素化相關蛋白。所以作者推測SNHG7可能通過招募CUL4A調控PTEN的泛素化。FISH-IF共染證明了CUL4A和SNHG7之間的共定位(圖6F),RIP和RNA pulldown證實了兩者間的結合關系(圖6G-H)。此外,co-IP實驗結果表明PTEN可以被CUL4A富集沉淀,而這種富集在過表達SNHG7之后顯著上調。以上表明SNHG7是通過招募CUL4A調控PTEN的泛素化。

圖6 SNHG7通過招募CUL4A促進PTEN泛素化激活PI3K/AKT通路
綜上所述,本研究表明外泌體SNHG7通過誘導LUAD細胞自噬和促進巨噬細胞M2極化促進LUAD細胞多西他賽耐藥。機制上,SNHG7招募HuR穩定ATG5和ATG12,誘導LUAD細胞自噬。同時,耐多西他賽的LUAD細胞分泌外泌體SNHG7,促進親代LUAD細胞耐多西他賽。此外,外泌體SNHG7通過招募CUL4A促進PTEN泛素化,激活PI3K/AKT通路,觸發巨噬細胞M2極化。

圖7 SNHG7誘導LUAD細胞多西他賽耐藥和巨噬細胞M2極化的分子機制
參考文獻:
Zhang Kai., Chen Jing., Li Chen., Yuan Yuan., Fang Surong., Liu Wenfei., Qian Yingying., Ma Jiyong., Chang Ligong., Chen Feifei., Yang Zhenhua., Gu Wei.(2021). Exosome-mediated transfer of SNHG7 enhances docetaxel resistance in lung adenocarcinoma. Cancer Lett, undefined (undefined), undefined. doi:10.1016/j.canlet.2021.10.029