






心臟成纖維細胞通過Htra3-TGF-β-IGFBP7軸調控心力衰竭的發生發展
組織纖維化和器官功能障礙是包括心力衰竭在內的與年齡相關的疾病的特征,但是否有一個共同的途徑來誘發這兩種事件仍不清楚。假設心臟成纖維細胞和心肌細胞之間可能存在通信,作者利用單細胞RNA-seq繪制的心臟細胞-細胞通訊圖。本文于2022年3月發表于《Nature communications》,IF= 12.121。
本文技術路線:

本文主要內容:
1、 單細胞網絡分析確定Htra3是心臟成纖維細胞的中心分子
為了研究導致衰竭心肌細胞誘導的分子相互作用,在進行主動脈橫向收縮術(TAC)或假手術的小鼠中,于2周后分離了心肌細胞和非心肌細胞(Fig. 1A)。利用所有數據集中的1019個配體和受體(LR)表達譜,作者生成了一個共表達網絡圖,并揭示了心臟中LR的細胞類型特異性轉錄特性 (Fig. 1B)。接下來,使用LR相互作用數據庫重新構建心臟LR相互作用網絡圖,并顯示相同或不同細胞類型之間潛在的綜合相互作用(Fig.1C)。通路分析顯示,PI3K-Akt、Rap1和TGF-β信號通路等特異性信號通路在心臟中被強烈激活(Fig.1d)。在各種細胞類型中,在包括心臟成纖維細胞與包括心肌細胞在內的多種細胞類型的相互作用最強(Fig.1e)。TAC引起的超負荷的壓力增加心臟成纖維細胞數量,其中細胞外基質相關基因如Ctgf、Tgfb1和Fbn1被激活(Fig.1f)。
通過生成心臟成纖維細胞網絡,作者確定Htra3是位于網絡中心的分子(Fig.1g)。Htra3的表達與心臟成纖維細胞模塊的表達有很強的相關性(Fig.1h),提示Htra3心臟成纖維細胞的身份。但其在心臟中的作用尚不清楚。心臟成纖維細胞特異性Htra3表達經scRNA-seq證實(Fig. 1j)。另外,作者也確認了HTRA3在人心臟間質中的表達(Fig. 1k)。
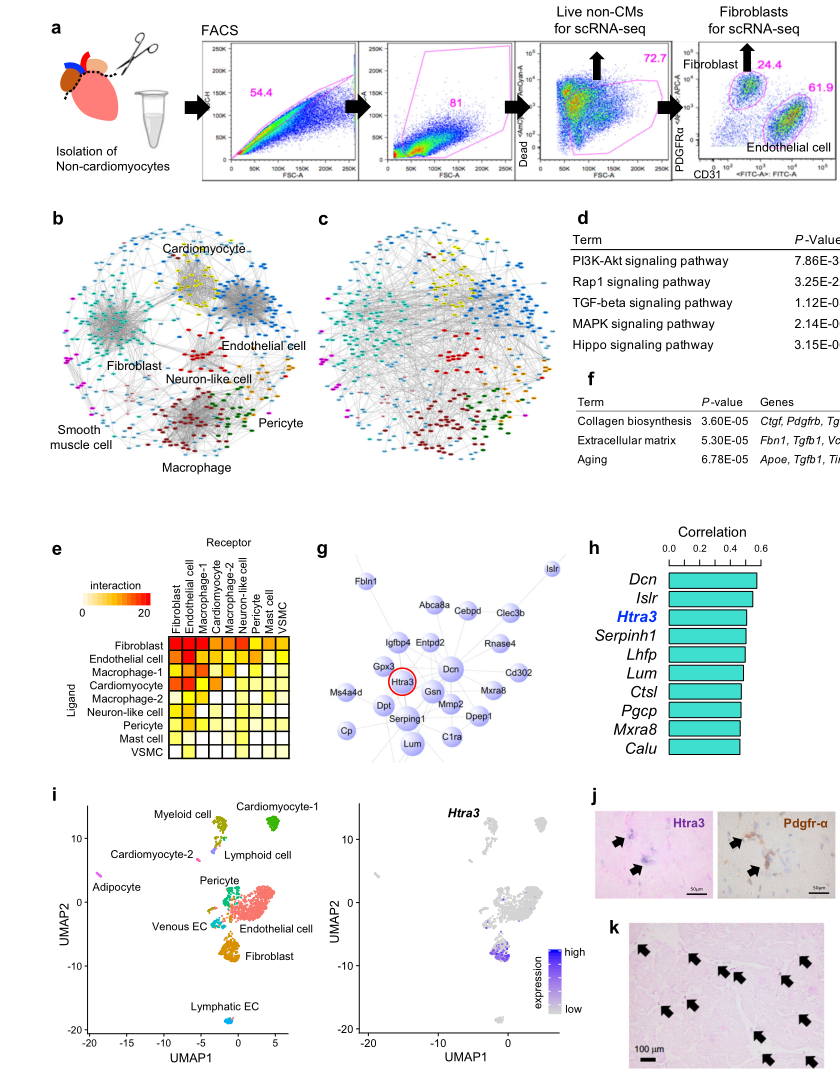
Fig1 單細胞網絡分析鑒定心臟成纖維細胞Htra3
2、心臟成纖維細胞Htra3支配心臟穩態,并因壓力過載而下調
與野生型相比,對照組小鼠,Htra3 KO小鼠即使在沒有壓力過載的情況下也表現出心肌肥厚,心肌細胞體積增大,而血壓沒有變化(Fig 2a-c)。TAC引起的輕微壓力過載可迅速誘導Htra3 KO小鼠嚴重心力衰竭,但在WT小鼠中不明顯(Fig 2b)。TAC對Htra3 KO小鼠的引起的心肌纖維化比WT小鼠嚴重(Fig 2d)。壓力過載降低了WT小鼠心肌成纖維細胞中的Htra3(Fig 2e)。人類心臟的scRNA-seq證實HTRA3在心臟成纖維細胞中特異表達,在衰竭心臟中表達減少(Fig 2f)。機械拉伸對分離的原代心臟成纖維細胞中抑制tra3和Tgfb1表達增加(Fig 2g)。這些結果表明,Htra3特異表達于心臟成纖維細胞,對于維持心臟基本大小和抵抗血流動力學過載至關重要,而心臟成纖維細胞的機械應激會降低Htra3的表達。

Fig2 Htra3 KO小鼠心肌及心肌細胞肥大,易受機械應激影響
3. htra3誘導TGF-β降解對預防心力衰竭和纖維化至關重要
在TAC引起的輕微壓力過載小鼠或假手術的WT和Htra3 KO小鼠中,于2周后對使用Pdgfr-α抗體分離的心臟成纖維細胞進行全長scRNA-seq(Fig 1a)。UMAP繪圖和基于圖形的聚類將心肌成纖維細胞分成三個聚類(C1-3),假時間分析顯示兩條軌跡(Fig 3a,b)。成纖維細胞軌跡1(來自C1 - C2)主要是由Htra3缺失引起,而成纖維細胞軌跡2 (C1 - C3)是由壓力過載引起的,Htra3缺失或不缺失均可 (Fig. 3a-c)。共表達網絡分析和隨機分析明確了M1和M17模塊顯著參與細胞分類(它們的模塊活性相互排斥(Fig. 3d)。M1模塊在心臟成纖維細胞中表達,包含先天免疫和toll樣受體信號通路相關基因(包括Tlr2和Tlr4)(Fig 3e, f)。軌跡1和軌跡2中M1模塊的活性被強烈抑制(Fig 3e-g)。
M17模塊富含參與細胞外基質形成和TGF-β信號通路的基因,它們的活性在兩個軌跡1和2中都有不同程度的上調 (Fig. 3h-j)。無論是壓力過載,還是Htra3缺失激活M17,它們一起協同增強其活性(Fig. 3j)。膠原原纖維組織基因Col1a1和Col3a1的表達在兩種軌跡中都被激活,而TGF-β信號分子的表達,如Tgfb3和Tgfbr2,以及活化心臟成纖維細胞6的標志物Postn,僅在軌跡2中被激活(Fig. 3i)。這些結果表明Htra3基本抑制TGF-β信號,壓力過載和Htra3缺失協同激活TGF-β信號,導致成纖維細胞活化。
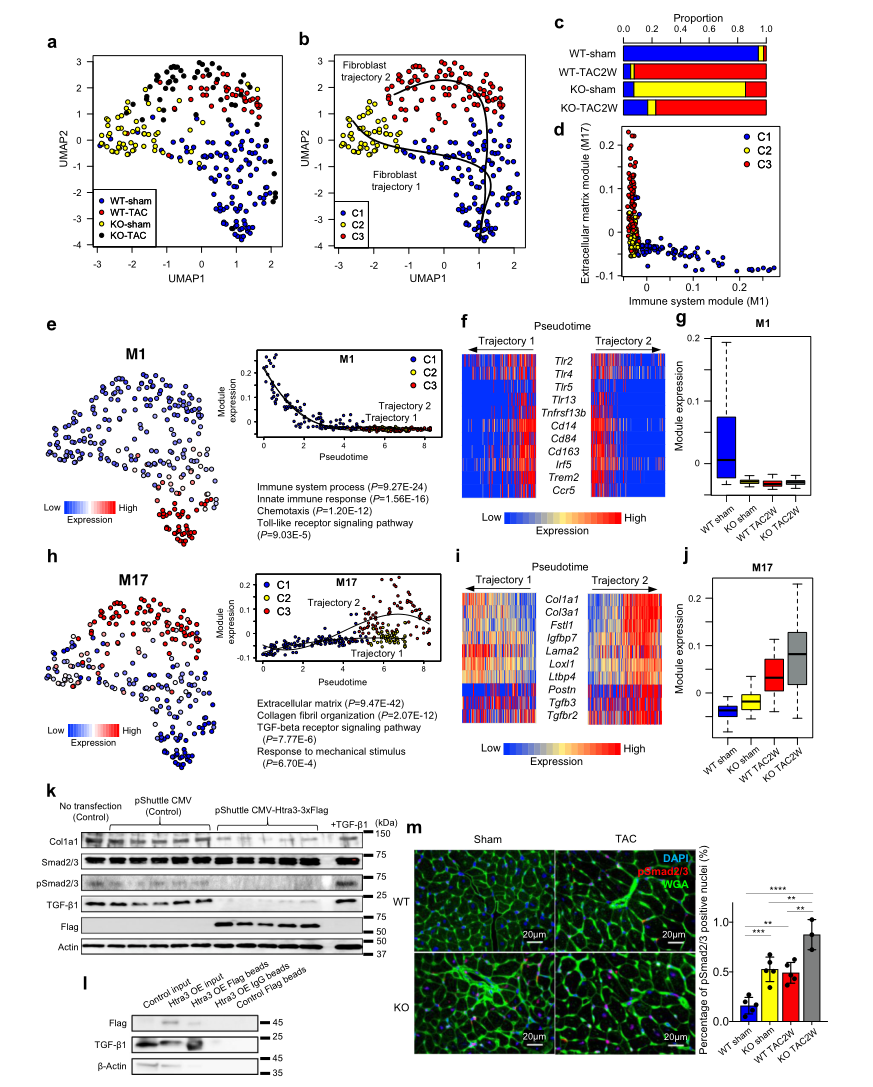
Fig3心臟成纖維細胞的單細胞rna序列顯示Htra3通過TGF-β抑制維持其靜止狀態
4. Htra3抑制誘導的TGF-β信號激活促進衰老衰竭心肌細胞的誘導
為了了解TGF-β信號的激活如何影響心肌細胞并導致心力衰竭,作者在TAC輕微壓力過載小鼠或假手術小鼠后在2周后對WT和Htra3 KO小鼠心肌細胞進行了scRNA-seq檢測。UMAP將心肌細胞劃分為4個簇,假時間分析顯示2條軌跡(Fig 4a, b)。共表達網絡分析和隨機森林分析識別模塊M1和M2在細胞分類中起重要作用。Htra3缺失,TAC輕微壓力過載,以及它們的聯合誘導了C1心肌細胞分別轉向C2, C3和C4(Fig. 4b)。富含線粒體氧化磷酸化和DNA修復相關基因(如Atm、Parp1/2和Rpa2)的M1模塊在軌跡1和軌跡2中均被顯著抑制(Fig 4d - f),這表明激活TGF-β信號可以抑制線粒體和DNA修復相關基因的表達(Fig 4d, e)。在Htra3 KO小鼠心肌細胞中,參與氧化還原過程的基因被下調,在無壓力過載的情況下,蛋白質合成相關基因表達上調, 可能導致htra3ko小鼠心肌細胞肥大 (Fig. 2c)。
模塊M2,包含與GTPase小信號(如Rhob和Rac1)、TGF-β受體信號(如Ltbp4和Tgfbr2)和p53信號(如Trp53和Cdkn1a),在C4中通過軌跡2特異激活 (Fig. 4g–i)。Htra3缺失和壓力過載的聯合作用顯著促進了用抗γH2A抗體染色的衰竭心肌細胞的誘導(Fig. 4j)。作者還證實了TAC手術后,Htra3 KO小鼠心肌細胞DNA損傷相關基因表達增加(Fig.4k)。M2還含有多種分泌因子,包括Bmp1、Cxcl12、Igfbp7,提示表達M2的心肌細胞表現出DNA損傷誘導的分泌表型,類似于衰老相關的分泌表型(SASP)(Fig 4l)。聚類分析發現,模塊M9富含編碼膠原纖維組織相關蛋白的基因(如Col1a1, Col3a1和Fbn1)和TGF-β信號傳導(如Tgfb3, Nox4, 和Postn),也像M2一樣在C4中被特異性激活 (Fig. 4m-o)。
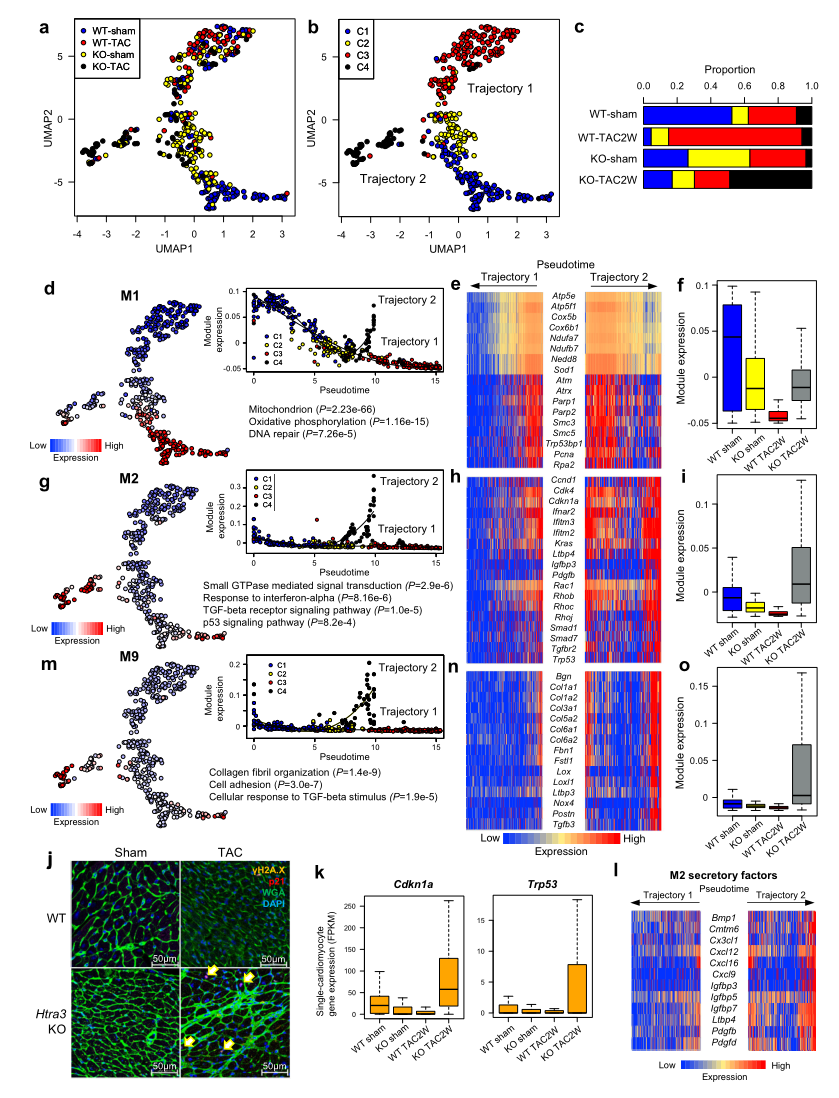
Fig4 心肌細胞的單細胞RNA-seq表明,Htra3可以防止心肌細胞衰老衰竭的誘導
5. TGF-β誘導的Nox4表達及隨后的p53激活對誘導具有分泌表型的衰竭心肌細胞至關重要
作者注射了Nox4- shRNA腺相關病毒9 (AAV9)載體來抑制Nox4的表達(Fig 5a)。超聲心動圖顯示,在TAC手術后htra3ko小鼠中,敲除Nox4可挽救其心功能障礙(Fig 5b)。免疫染色和western blot分析顯示,Nox4抑制可顯著降低DNA損傷(Fig 5c, d)和TGF-β信號蛋白相關分子的表達(Fig 5e)。作者在TAC手術后1周將AAV9-Htra3載體注入WT小鼠體內。發現心臟Htra3過表達抑制TGF-β信號傳導,改善心功能障礙和纖維化。
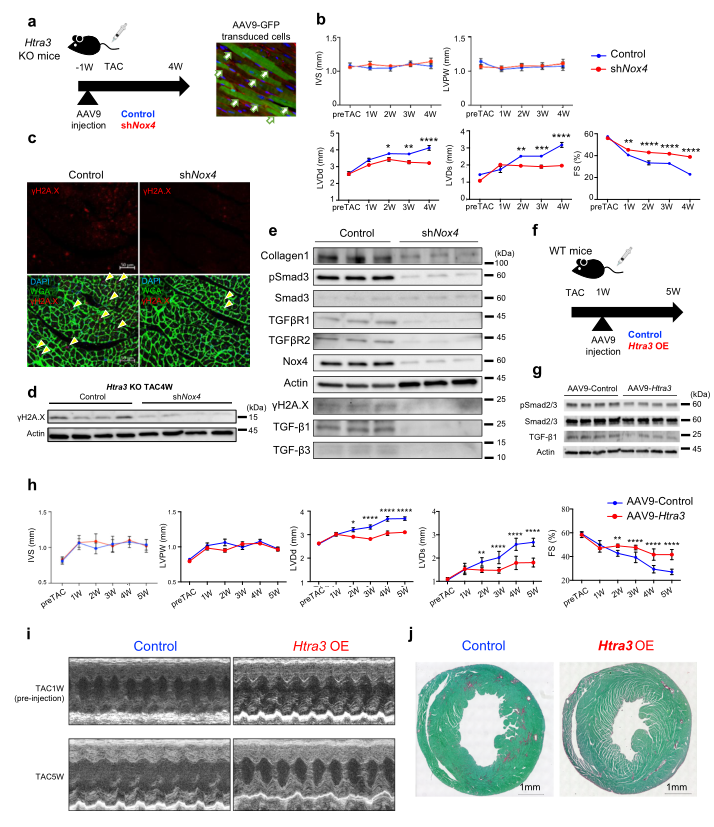
Fig5 TGF-β誘導的Nox4激活可誘導衰老衰竭心肌細胞和心力衰竭
6、空間轉錄組顯示Htra3-TGF-β軸在心肌梗死后心肌纖維化和心肌細胞分泌表型誘導中起重要作用
為了闡明Htra3如何在空間上調控心臟重構,作者研究了Htra3在心肌梗死(MI)模型中的作用。Visium (10X Genomics)的空間轉錄組分析顯示心肌梗死后梗死區Htra3高表達,提示心肌重構可能存在空間調控(Fig 6a)。與WT小鼠相比,Htra3 KO小鼠心肌梗死后出現嚴重的心臟重構(Fig 6b)。超聲心動圖分析顯示Htra3KO小鼠表現出明顯的心臟擴張和收縮功能障礙 (Fig 6c)。通過對野生型和Htra3 KO小鼠假手術或MI手術后心臟組織的空間轉錄組分析,發現了具有空間定位特征的特定基因集群(Fig.6d)和特異性表達譜Htra3(Fig. 6e)。MI誘導了線粒體中簇1基因的表達 (Fig. 6d, e)。Htra3 KO小鼠心肌梗死后,與梗死區對應的簇2區域擴大(Fig. 6d)。利用scRNA-seq圖譜對空間定位點進行反螺旋分析,表明成纖維細胞在聚類2的區域高度富集(Fig. 6f)。作者還利用scRNA-seq譜對MI后心肌細胞進行了軌跡分析,發現MI誘導的心肌細胞以TGF-β信號相關分子和分泌因子的表達為特征 (Fig. 6g, h)。空間轉錄組分析顯示,這些基因與細胞外基質基因在心肌梗死后Htra3 KO小鼠的梗死區特異激活 (Fig. 6i)。提示Htra3下調可促進心肌纖維化,誘導梗死區心肌細胞分泌表型。
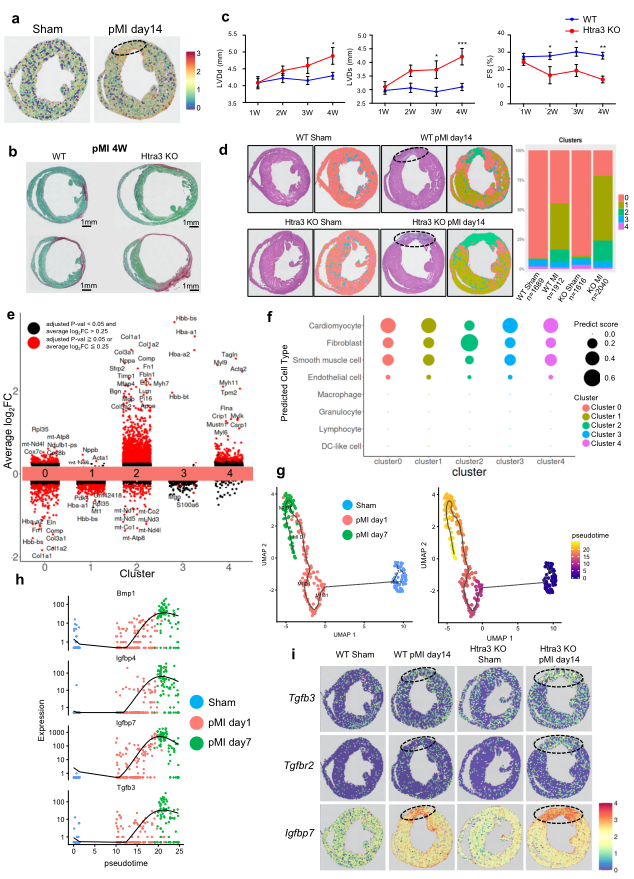
Fig6 空間轉錄組分析揭示Htra3抑制介導的衰老衰竭心肌細胞空間誘導
7、 沉默外泌體來源的circTRPS1通過GLS1調控抑制了BCa在體內的生長和轉移
UMAP繪圖和基于圖形的聚類將心肌細胞分為3個聚類,假時間分析顯示2條軌跡(Fig 7a, b)。軌跡1和軌跡2分別對應C1到C2和C1到C3的過渡(Fig7b)。對照組心肌細胞主要屬于C1,心衰患者心肌細胞主要屬于C2或C3(Fig 7c)。M2富含與線粒體電子傳輸和ATP生物合成有關的基因(Fig 7d),在兩種軌跡中都受到抑制 (Fig. 7d–f)。DNA損傷反應(CDKN1A、MDM4和RBX1)在軌跡中特異激活M2(Fig7g-i),其中M1和M2在模塊活動中表現出互斥關系 (Fig. 7j)。M1還含有特異性表達于小鼠衰老心肌細胞的分泌因子(Fig.7h)。此外,M44與細胞外基質組織相關的基因富集,并在軌跡2中特異激活 (Fig. 7k-m)。衰老衰竭心肌細胞表達的細胞因子TGF-β3、LTBP4和IGFBP7的水平在心力衰竭患者中較高(取決于嚴重程度),移植后較低,類似于NT-proBNP(一種成熟的心力衰竭生物標志物)(Fig. 7n)。隨機森林分析表明,IGFBP7蛋白在非晚期心力衰竭和晚期心力衰竭的分類中起著最重要的作用(Fig.7o)。受體工作特征分析顯示NT-proBNP有更好的診斷心力衰竭的能力,而IGFBP7有更高的能力來判斷心力衰竭的嚴重程度(Fig.7p, q)。這些結果表明,從衰竭的心肌細胞分泌的IGFBP7是一種有用的人類晚期心力衰竭的生物標志物。
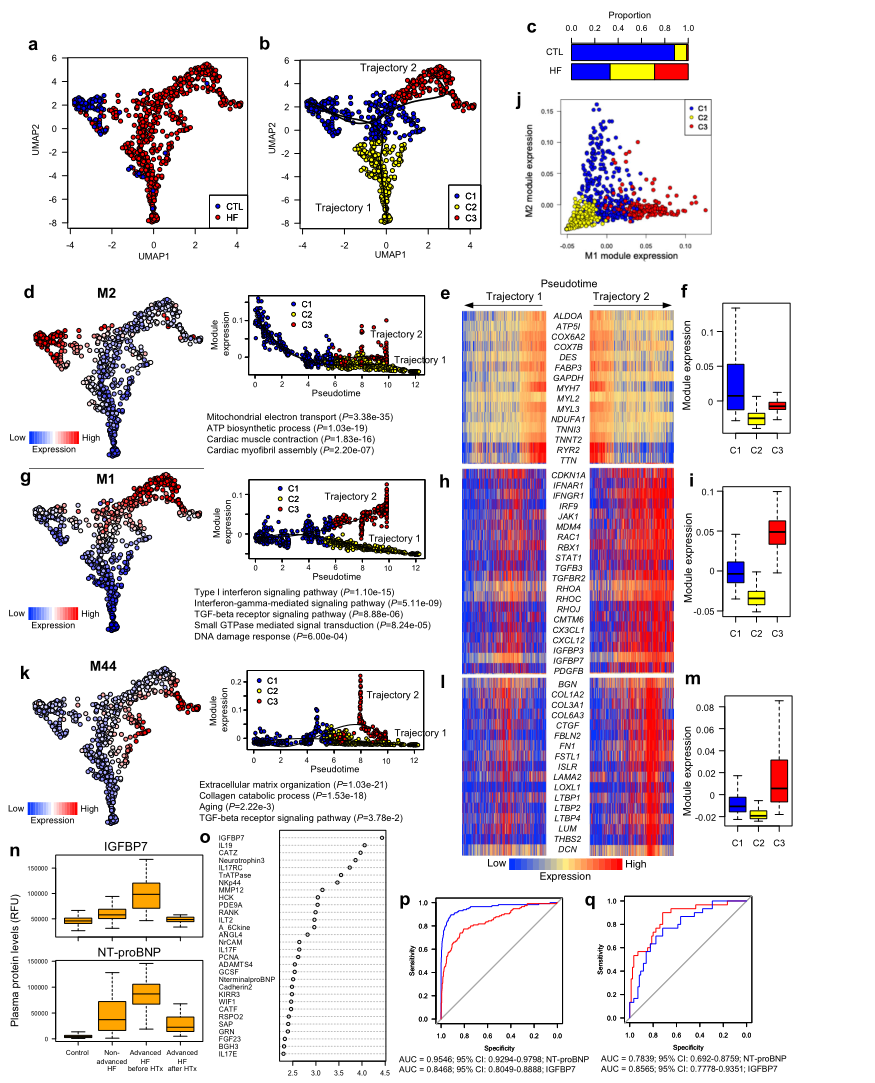
Fig7 心衰患者的單心肌細胞RNA-seq和血漿蛋白組分析
綜上所述,本文發現了一些由衰竭心肌細胞分泌的與心力衰竭嚴重程度相關的分泌因子。作者證明了心臟成纖維細胞通過誘導不僅在心臟衰竭的發展中發揮重要作用,心肌細胞也通過Htra3-TGF-β-IGFBP7軸分泌表型。有利于推動有關心力衰竭發病機制的進一步研究。
參考文獻:
Ko, T., et al., Cardiac fibroblasts regulate the development of heart failure via Htra3-TGF-beta-IGFBP7 axis. Nat Commun, 2022. 13(1): p. 3275.