






胃癌中METTL16乳酸化通過FDX1 mRNA的m6A修飾促進細胞凋亡

銅是生物體必需的微量元素,參與細胞生長和代謝。銅穩態失調與多種腫瘤相關,癌癥患者血清銅水平升高與癌癥分級和化療耐藥相關。如果銅的濃度超過一定的閾值,就會產生毒性,導致一種新發現的細胞死亡形式,即銅中毒。當銅直接與三羧酸循環的脂基化組分結合時,就會發生銅死亡,導致隨后含鐵-硫簇的蛋白質喪失、蛋白毒性應激并最終導致細胞死亡。鐵氧化還原蛋白1(FDX1)編碼一種還原酶,可將Cu2+還原為毒性更強的Cu1+;硫辛酸二氫硫辛酰胺S-乙酰轉移酶(DLAT)是銅中毒所必需的酶。然而,細胞凋亡在腫瘤中的啟動、傳播和執行機制仍不清楚。m6A是真核細胞中最豐富的RNA修飾,在腫瘤發生中起著重要的作用。METTL16在多種腫瘤中以m6A依賴的方式表現出腫瘤發生和促進腫瘤的能力,但控制METTL16活性的調節機制尚不清楚。乳酸源性乳糖化是新近發現的一種翻譯后修飾(PTM)。乳酸被腫瘤細胞攝取并轉運到線粒體進行氧化提供能量,同時衍生組蛋白賴氨酸殘基的乳糖化來刺激基因轉錄。組蛋白乳糖化修飾已被證明涉及多個病理過程,如巨噬細胞極化和腫瘤發生。細胞中非組蛋白的豐度和特異性仍高于組蛋白。非組蛋白是否存在大量的乳糖化修飾,以及這些乳糖化的非組蛋白如何在腫瘤進展中發揮作用和被調節,這些都亟待探索。該研究發表在《Nature Communications》,IF:16.6。
技術路線

結果
1. 胃癌組織中銅含量較高,且與腫瘤進展相關
銅死亡是由銅濃度過高引起的,但銅死亡在腫瘤中的調控機制仍需探索。由于銅主要通過胃和小腸上部吸收,因此胃腸道系統的腫瘤適合研究銅突的發生和傳播。研究者分析了48對胃癌組織和癌旁組織中的銅濃度,發現胃癌組織中的銅濃度高于正常胃組織(圖1a)。進一步分析發現,ⅲ期胃癌患者的相對銅含量高于ⅰ期和ⅱ期胃癌患者(圖1b),表明銅含量與腫瘤進展相關。此外,銅含量與胃癌患者的總生存期(OS)和無病生存期(DFS)呈負相關(圖1c, d)。同時,銅含量與細胞增殖指標Ki-67表達呈正相關(圖1e)。同樣,銅含量與患者血清標本中的血小板/淋巴細胞和中性粒細胞/淋巴細胞比值呈正相關,兩者都是與炎癥相關的胃癌侵襲性惡性腫瘤的預后生物標志物(圖1f, g)。這些結果表明,銅的高含量參與了氣相色譜的發生和發展。
此外,研究者還檢測了銅在不同GC類型中的濃度差異,尤其是在惡性腫瘤中。有趣的是,研究者發現黏液腺癌中的銅濃度高于整個胃腫瘤(圖1h)。黏液腺癌是胃癌的一種罕見組織學亞型,與非黏液腺癌相比,黏液腺癌的分期更晚,5年OS和DFS更差。黏液腺癌作為一種難治性胃癌,具有放化療抵抗的特點,迫切需要更有效的治療方法。
最近,研究人員發現,細胞內的高銅濃度觸發了還原酶FDX1,將Cu2+還原為毒性更強的Cu1+,導致銅死亡。研究者發現FDX1在胃癌中的蛋白水平高于正常胃組織(圖1k)。由于FDX1蛋白水平和銅含量在GC中都較高,因此可能更容易觸發銅突。這為胃癌尤其是惡性腫瘤——黏液腺癌提供了潛在的治療策略。
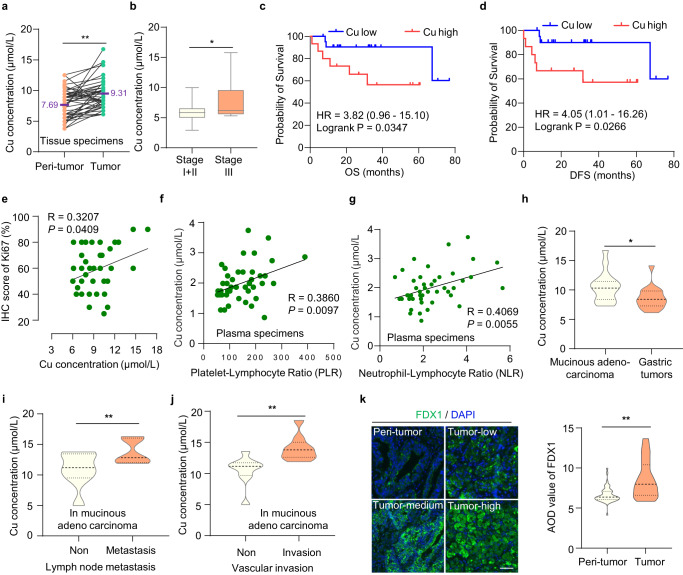
圖1 胃癌組織中銅含量較高,且與腫瘤進展相關
2. METTL16是銅死亡的關鍵介導因子
有研究表明,與良性胃病患者和健康對照患者相比,胃癌患者的總m6A水平顯著升高,并促進胃癌的發生、生長、侵襲、上皮間質轉化、轉移甚至多藥耐藥的過程。此外,m6A修飾還參與多種細胞死亡類型,包括凋亡、壞死性凋亡、鐵死亡和焦亡。然而,m6A修飾與銅死亡的關系尚不清楚。有趣的是,研究者發現銅含量與GC組織中總m6A水平呈正相關(圖2a)。此外,銅顯著上調胃癌細胞中m6A修飾的總水平(圖2b)。鑒于甲基轉移酶樣蛋白(METTL)家族在促進m6A修飾中起主導作用,研究者分析了以1:1的比例用來來洛莫/雙硫侖和銅處理METTL敲低的細胞時的活力。結果表明,METTL16敲低導致對來來洛莫/雙硫侖-銅處理的耐藥性(圖2c-e)。此外,在對照組和METTL16敲低的細胞中,四硫鉬酸鹽處理均抑制了銅綠假單胞菌脫除,表明METTL16參與了銅綠假單胞菌脫除(圖2f)。為了進一步驗證這些結果,研究者成功構建并驗證了METTL16敲除克隆細胞系,觀察到與METTL16敲除細胞相似的結果(圖2g, h)。這些結果表明,METTL16在銅突病中起著至關重要的作用。
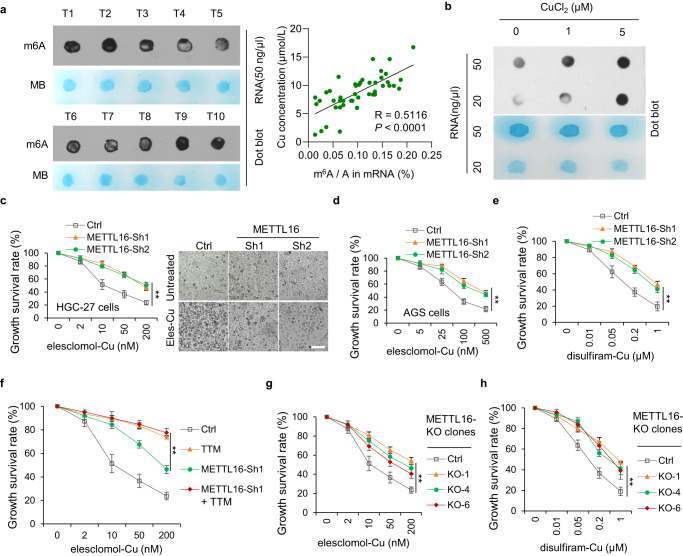
圖2 METTL16是銅死亡的關鍵介導因子
3. METTL16通過靶向FDX1促進銅死亡
METTL16在許多轉錄本中負責m6A的沉積。因此,研究者在穩定的Ctrl-Sh和METTL16-Sh細胞系中進行了甲基化RNA免疫沉淀(MeRIP)測序(圖3a)。使用HOMER軟件預測每個樣本中潛在的m6A修飾,m6A修飾主要映射到經典的GGAC基序(圖3b)。根據MeRIP-seq結果,研究者進一步分析了mRNA的總m6A分布模式,發現m6A峰主要富集在CDS和3'UTR區域。GO富集分析顯示METTL16修飾基因富集的前15條通路,其中部分與線粒體通路相關(圖3c)。此外,研究者從METTL16-sh組中鑒定出10個典型的與m6A甲基化水平低以及mRNA水平下調相關的因子(圖3d)。qPCR分析顯示,在METTL16-Sh細胞中,FDX1、MTF1、PDHB、ATP7A和DLD的mRNA水平降低。與對照細胞相比,FDX1在METTL16-sh細胞中表現出最顯著的mRNA水平降低(圖3e)。此外,在METTL16-Sh或-KO細胞中檢測到FDX1的蛋白水平顯著降低(圖3f, g)。考慮到FDX1是銅離子載體誘導的細胞死亡的關鍵調節因子,研究者推測METTL16誘導的細胞凋亡是FDX1依賴的。
接下來,研究者評估了METTL16是否通過影響FDX1的表達來調節細胞凋亡。鑒于蛋白質脂化是銅胞嘧啶的關鍵因素,研究者使用硫辛酸特異性抗體作為DLAT脂化的指標,評估了METTL16敲低是否影響蛋白質脂化。結果表明,如免疫印跡測定的那樣,METTL16敲低導致DLAT脂化降低(圖3h)。此外,FDX1過表達逆轉了在來來洛莫/雙硫侖-cu處理后,METTL16敲低和敲除細胞系中銅突亡的拮抗作用(圖3i, j)。FDX1敲低可抑制METTL16過表達誘導的銅突(圖3k)。綜上所述,這些數據表明METTL16通過靶向FDX1促進銅死亡。
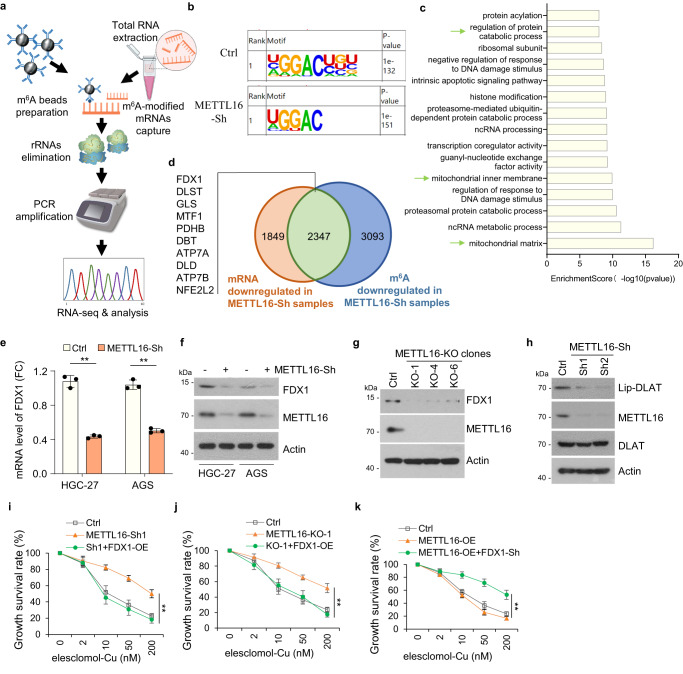
圖3 METTL16通過靶向FDX1促進銅突
4. METTL16通過對FDX1 mRNA的m6A修飾促進FDX1的積累
為了進一步探索METTL16如何調控m6A修飾進而影響FDX1 mRNA水平,研究者進行了基因特異性MeRIP-qPCR檢測。研究者發現,在METTL16敲低和敲除細胞中,FDX1 mRNA上的m6A水平顯著降低,而在轉染METTL16過表達質粒的細胞中,FDX1 mRNA上的m6A水平顯著升高(圖4a, b)。當用ActD停止轉錄時,METTL16敲低細胞中FDX1 mRNA的衰減速率比對照細胞快(圖4c)。這些結果表明,METTL16介導的甲基化導致FDX1 mRNA的穩定性。對FDX1富集的m6A峰的綜合基因組學觀察(IGV)分析顯示,與對照細胞相比,METTL16-sh細胞中的m6A水平降低,表明METTL16可能促進FDX1在CDS或3'UTR區域的m6A修飾(圖4d)。因此,研究者將FDX1的部分CDS和3'UTR區融合到pGL3熒光素酶報告基因的下游,構建FDX1-WT熒光素酶報告基因。共轉染FDX1熒光素酶報告基因和不同劑量的METTL16-OE質粒,研究者發現METTL16顯著促進FDX1熒光素酶的活性(圖4e)。
為了進一步研究,研究者使用基于序列的m6A修飾位點預測器SRAMP預測了FDX1 mRNA上可能的m6A修飾位點。綜合圖4e中的預測結果和數據,研究者確定了兩個潛在的m6A修飾位點,它們的置信度非常高:位于CDS區域的位點602a和位于FDX1轉錄本UTR區域的位點820 A(圖4f, g)。接下來用特異性引物進行的MeRIP-qPCR分析證明,FDX1轉錄本上的602位點是METTL16介導的甲基化的直接底物(圖4h)。此外,研究者基于FDX1-WT熒光素酶報告基因,通過將m6A基序中的特異性腺苷(A)替換為胸腺嘧啶(T),設計并構建了FDX1-602-Mut和FDX1-820-Mut熒光素酶報告基因(圖4g)。雙熒光素酶實驗結果表明METTL16不能促進602位點突變的FDX1-CDS/UTR報告載體的熒光素酶活性(圖4i)。GEPIA數據庫中METTL16和FDX1的表達分析表明,METTL16和FDX1在GC中呈正相關(圖4j)。此外,免疫組織化學染色顯示,在包含54對胃癌和癌旁正常組織的組織芯片中,METTL16和FDX1的表達呈正相關(圖4k)。綜上所述,這些結果證實了METTL16介導的FDX1 mRNA的m6A修飾對FDX1 mRNA的穩定性至關重要。
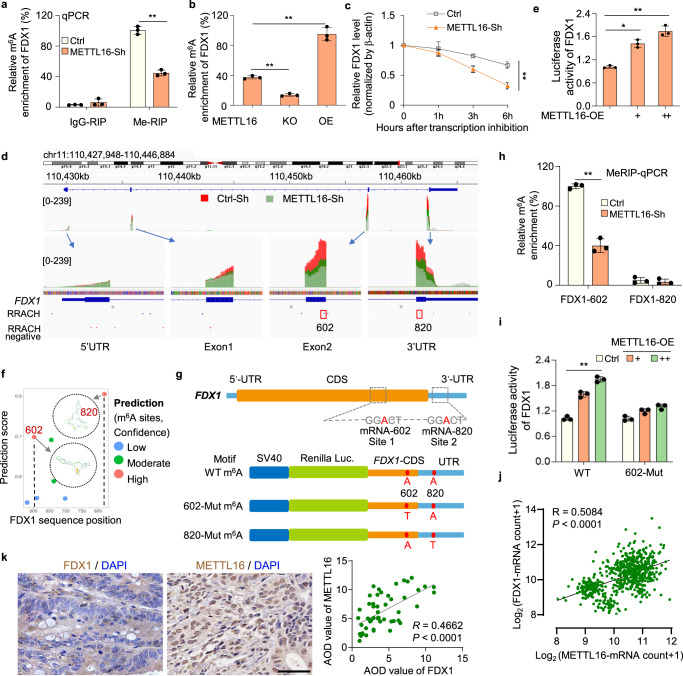
圖4 METTL16通過對FDX1 mRNA的m6A修飾促進FDX1的積累
5. 在銅脅迫下,METTL16在K229處發生乳酸化
接下來,研究者評估了METTL16是否對GC中的銅脅迫有響應。在銅處理后,METTL16的mRNA和蛋白水平沒有變化,但FDX1的蛋白水平顯著增加(圖5a, b)。為了驗證在銅脅迫下FDX1表達的上調是否依賴于METTL16,研究者檢測了在銅存在或不存在的情況下,METTL16敲低或敲除細胞中FDX1蛋白的表達。結果表明,METTL16缺陷消除了銅誘導的FDX1上調(圖5c, d)。由于METTL16的mRNA和蛋白水平在銅脅迫下沒有變化,研究者隨后使用質譜檢測了METTL16的PTM的變化。分析顯示METTL16蛋白序列上有11個乳糖化位點和2個乙酰化位點(圖5e)。在乳酸處理后,6個乳糖化位點的PTM水平顯著升高,這意味著這6個位點在腫瘤細胞中可能受到調控。然后,研究者證實了銅脅迫下存在乳糖化或乙酰化反應。結果表明,銅處理顯著增加了METTL16的乳糖化水平而不是乙酰化水平(圖5f)。為了確定METTL16在銅相關代謝中重要的乳化位點,研究者產生了這6個乳化位點的乳化缺陷突變體(K→R)和K410。研究者發現,只有METTL16-K229R突變體在銅脅迫下顯示出乳糖化降低(圖5g)。隨后,LC-MS/MS分析顯示METTL16-K229與其他乳糖化位點的b-y離子匹配圖見圖5e(圖5h)。為了進一步證實銅脅迫下METTL16-K229的乳糖化,研究者制備了METTL16 lacty-K229抗體,并利用lacty-K229肽段和表達METTL16-WT、-K229R或-K229E的細胞驗證了其有效性和特異性(圖5i, j)。銅和乳酸處理均誘導K229處METTL16的強乳糖化(圖5k, l),表明K229是銅相關代謝中必不可少的乳糖化位點。這些結果表明,在銅脅迫下,METTL16在K229發生乳酸化。
在穩定的METTL16挽救細胞系中,FDX1蛋白水平進一步得到保證,其中內源性METTL16被野生型(WT)、乳酸化缺陷型(K229R)或乳酸化模擬型(K229E)取代。結果顯示,FDX1蛋白水平在METTL16 -敲低的細胞中降低,在METTL16-WT或-K229E拯救細胞后恢復,但在METTL16-K229R拯救細胞后沒有恢復(圖5m)。銅脅迫誘導METTL16-K229乳糖化,促進METTL16甲基轉移酶活性。
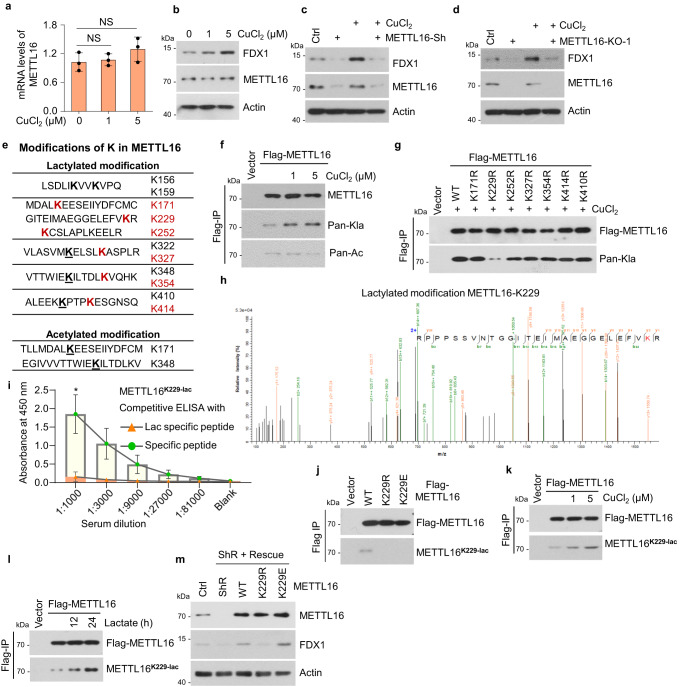
圖5 在銅脅迫下,METTL16在K229處發生乳酸化
6. SIRT2使METTL16- k229脫酰化并抑制METTL16活性
上述數據證實銅激活METTL16-K229的乳糖化,但具體參與腫瘤細胞的乳糖化酶和乳糖化轉移酶尚不清楚。研究者研究了銅是否介導了對METTL16的乳糖基轉移酶或脫乳糖基酶的調節。SIRT2被鑒定為METTL16的結合蛋白,是一種典型的去乙酰化酶,分為主要的脫乙酰酶和乳糖基轉移酶組(圖6a)。EIF3b也被鑒定為METTL16的結合蛋白,這與之前的研究一致。為了研究和驗證METTL16和SIRT2之間的相互作用,研究者在HGC-27細胞中檢測到METTL16-SIRT2的結合(圖6b)。METTL16-SIRT2的直接關聯通過His-pull-down試驗得到驗證(圖6c)。SIRT2過表達顯著抑制METTL16-K229的乳糖化(圖6d),而SIRT2敲低或SIRT2抑制劑AGK2處理則促進METTL16-K229的乳糖化(圖6e)。此外,銅誘導的METTL16-K229的乳糖化被SIRT2過表達抑制,而被SIRT2敲低或AGK2處理增加(圖6f, g)。綜上所述,這些結果表明SIRT2在K229位點與METTL16相互作用并使其脫酰化。
研究者進一步探索了SIRT2對METTL16的調控作用在穩定的METTL16挽救細胞株中的作用。SIRT2過表達抑制了METTL16- wt細胞中的FDX1 m6A水平,但未抑制METTL16-K229r或-K229E細胞中的FDX1 m6A水平,表明SIRT2通過METTL16-K229的乳糖化抑制了METTL16的活性(圖6h, i)。此外,SIRT2過表達抑制FDX1蛋白水平(圖6j),而SIRT2敲低和AGK2處理促進FDX1蛋白水平(圖6k)。免疫熒光染色結果顯示,在胃癌組織芯片中,SIRT2蛋白水平與FDX1蛋白水平呈負相關(圖6l)。綜上所述,這些結果表明SIRT2通過在K229上去乙酰化來負調控METTL16的活性。
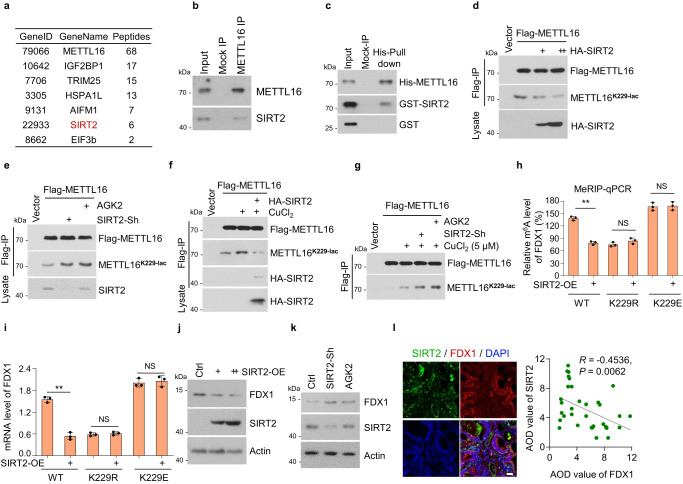
圖6 SIRT2使METTL16-K229脫酰化并抑制METTL16活性
7. SIRT2-METTL16-fdx1細胞凋亡軸支持一種有前景的治療策略
FDX1是銅死亡的重要介質,而銅死亡是癌癥的潛在治療方法。研究者發現,在SIRT2抑制或銅誘導METTL16-K229乳糖化后,FDX1蛋白水平升高。接下來,研究者研究了METTL16-K229乳糖化對銅突細胞的影響。結果表明,在銅脅迫下,METTL16敲低介導的DLAT脂化降低在METTL16-WT/-K229E細胞中被恢復,但在METTL16-k229r細胞中沒有恢復(圖7a)。此外,在銅存在的情況下,來來洛莫處理可誘導METTL16-WT或-K229E細胞發生銅中毒,但對METTL16-K229R細胞未發生銅中毒(圖7b)。此外,SIRT2過表達逆轉了在銅脅迫下由METTL16-WT引起的DLAT脂化的增加,而不是由METT16-K229E引起的DLAT脂化的增加,表明SIRT2通過去乙酰化METTL16-K229來抑制銅死亡(圖7c)。AGK2處理同樣促進了銅脅迫下METTL16-WT細胞的DLAT脂酰化,但對METTL16-K229R細胞沒有作用(圖7d)。此外,AGK2處理促進了在有來昔洛莫和銅存在的情況下的銅突(圖7e)。
研究者進一步評估了METTL16乳糖化是否可以增強接受來來洛莫治療的小鼠異種移植瘤的治療反應。將METTL16-WT、-K229R、-K229E細胞分別于裸鼠左右后肢上方皮下注射。在METTL16-WT和-K229E組中,接受來來洛莫治療的小鼠的腫瘤生長明顯受到抑制,而在METTL16-K229R組中,腫瘤的體積和重量減少(圖7f-h)。
腫瘤切片中的FDX1染色顯示,METTL16-WT和-K229E組的腫瘤切片中FDX1染色高于METTL16-K229R組(圖7i)。這些結果表明,METTL16在K229的泌乳狀態在決定銅突的發生中起著至關重要的作用。此外,在異種移植瘤中使用來來洛莫和AGK2聯合治療。對腫瘤切片的分析表明,AGK2在體內可致敏來來洛莫治療,如觀察到的腫瘤體積和重量的減少(圖7j-l)。這些結果表明,來來洛莫聯合AGK2治療胃癌,尤其是惡性腫瘤黏液腺癌是一種有前景的治療方法。
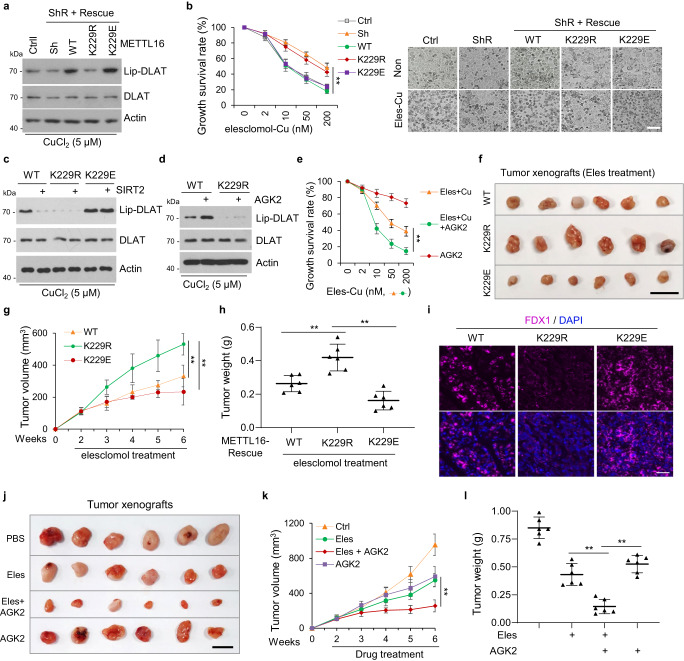
圖7 SIRT2 - METTL16 - fdx1細胞凋亡軸支持一種有前景的治療策略
結論
這些結果表明,METTL16是銅死亡的驅動力。METTL16在K229上的乳化通過對FDX1 mRNA的m6A修飾上調FDX1蛋白的表達,這是銅脅迫下觸發銅中毒的途徑之一。此外,研究結果表明METTL16在胃癌中是協調葡萄糖和銅代謝的中心樞紐。此外,結果表明METTL16的乳糖化為評價銅離子載體藥物的有效性提供了一個潛在的靶點。鑒于胃腫瘤比正常組織具有更高的銅和乳酸濃度,利用銅離子載體結合銅離子和SIRT2特異性抑制劑可能是一種可行的胃癌治療策略。
實驗方法
細胞培養,小鼠異種移植腫瘤實驗,m6斑點印跡測定,qRT-PCR,熒光素酶報告基因檢測,蛋白質印跡,His-pull-down測定,免疫沉淀,免疫熒光染色,MeRIP-qPCR,RNA穩定性檢測,體外乳酸化試驗,生物信息學分析
參考文獻
Sun L, Zhang Y, Yang B, Sun S, Zhang P, Luo Z, et al. Lactylation of METTL16 promotes cuproptosis via m6A-modification on FDX1 mRNA in gastric cancer. Nat Commun. 2023 Oct 20;14(1):6523. doi: 10.1038/s41467-023-42025-8.