






鐵死亡孔誘導Ca2+通量和ESCRT-III激活來調節細胞死亡動力學

鐵死亡是一種與脂質過氧化相關的調節壞死的鐵依賴形式。盡管它在鐵死亡的炎癥結果中起關鍵作用,但在這種類型的細胞死亡過程中導致質膜破壞的分子事件知之甚少。
2020年11月,來自德國科隆大學遺傳學研究所、埃伯哈德-卡爾大學生物化學學院間研究所、德拉薩大學化學系等團隊合作在Cell Death & Differentiation雜志上發表了文章“Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics.”。此文獻發現胞質Ca2+的持續增加是鐵死亡的一個標志,它發生在細胞完全破裂之前。質膜損傷導致鐵死亡與半徑只有幾納米的膜納米孔有關,而鐵死亡可以被滲透保護劑延遲,而不是脂質過氧化。鐵死亡過程中的Ca2+通量可誘導ESCRT-III 依賴的膜修復機制的激活,從而平衡細胞死亡動力學并調節鐵死亡的免疫學特征。團隊關于鐵死亡的發現提供了一個統一的概念,即在質膜破裂之前胞漿Ca2+的持續增加是受調控的壞死類型的共同特征,并將ESCRT-III激活作為這些溶性細胞死亡途徑中的一般保護機制。
鐵死亡是一種不依賴于caspase的調控性壞死,其特征是細胞膜中產生鐵依賴的脂質過氧化物。通過鐵死亡導致的細胞死亡的特征是質膜破裂和釋放其他受限的細胞內成分,包括促炎癥損傷相關的分子模式。因此,鐵死亡這種類型的細胞死亡與壞死性炎癥和先天免疫系統的激活有關。已有研究表明,鐵死亡可導致缺血/再灌注損傷、組織損傷和器官死亡,以及其他幾種病理,包括神經退行性疾病和癌癥。因此,了解鐵死亡過程中膜破裂的分子機制不僅具有生物學意義,而且具有醫學意義。
執行鐵死亡的一個基本步驟是質膜的最終破壞。然而,然而,導致質膜完整性喪失的分子機制以及鐵死亡癥膜損傷的性質和大小仍未被探索。其他形式的壞死細胞死亡,如壞死性死亡、或毒素誘導的細胞死亡對質膜的損傷導致離子通量的激活。在這些情況下,Ca2+通量與膜修復機制的激活有關。轉運所需的核內體分選復合體(ESCRT)機制似乎發揮了關鍵的平衡作用,可以延遲壞死和焦亡中的細胞死亡。
在不同的細胞模型中,研究了Erastin-1和RSL3觸發鐵死亡時質膜通透性的分子機制。利用活細胞成像和流式細胞術,我們并行追蹤了鐵死亡不同特征的動力學。我們發現在鐵死亡過程中脂質過氧化發生在胞質Ca2+持續增加和最終質膜破裂之前。我們還確定納米孔的形成是鐵死亡過程中觸發質膜破裂的核心機制,因此,適當大小的滲透保護劑可以抑制質膜破裂。最后,將鐵死亡中胞質Ca2+的增加與ESCRT-III機制的激活聯系起來,該機制作為一種延遲細胞死亡的保護機制。這對免疫有影響,因為ESCRT-III的缺失可調節鐵死亡細胞的細胞因子分泌,從而重塑鐵死亡的炎癥特征。研究結果支持ESCRT-III機制在調節壞死過程中平衡膜損傷和調節免疫結果的一般作用。
技術路線:
一、胞漿內Ca2+的持續增加是鐵死亡的一個標志
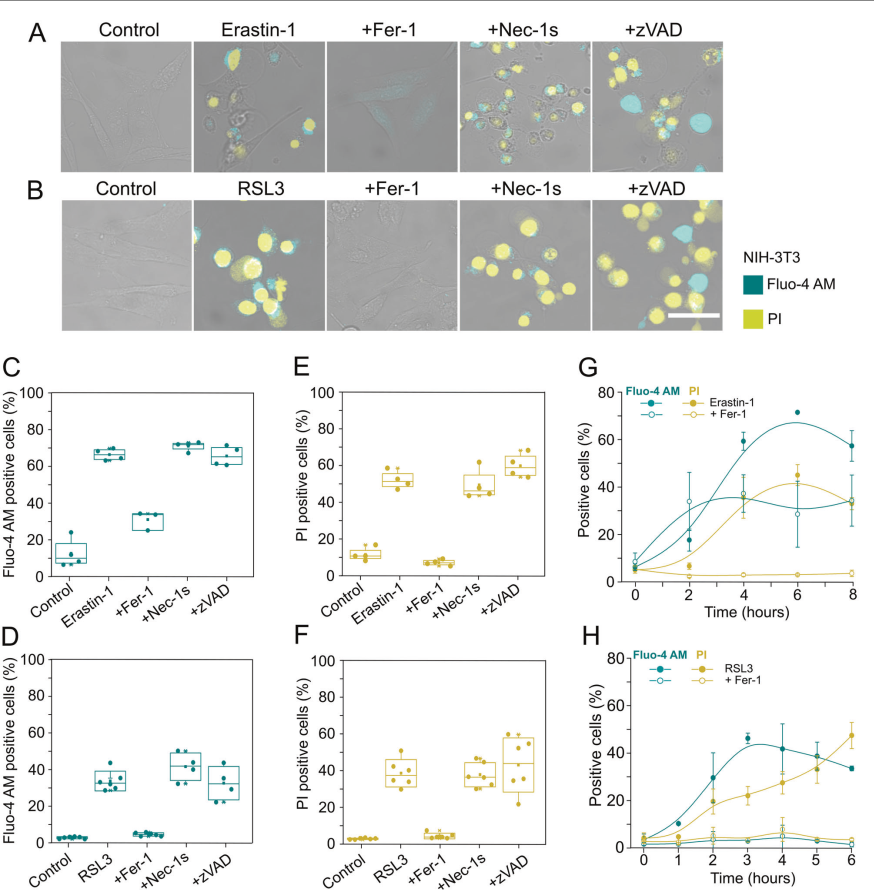
在使用Erastin-1或RSL3處理的小鼠成纖維細胞(NIH-3T3)中,我們通過活細胞共聚焦成像(圖1A, B)和流式細胞術(圖1C H)檢測細胞漿Ca2+水平,同時檢測細胞形態和質膜破裂的變化。我們使用氟-4乙酰氧基甲基(氟-4 AM)作為Ca2+指示劑[27]的熒光形式,來觀察細胞內Ca2+水平,PI作為不可逆質膜破壞和細胞死亡的標記。
在使用Erastin-1或RSL3處理NIH-3T3細胞后,胞漿Ca2+濃度明顯升高(圖1A, B)。這伴隨著細胞形狀的改變,包括細胞圓形和出現單個腫脹水泡,隨后質膜完整性完全破裂。當用RSL3處理人類纖維肉瘤細胞(HT-1080)和人類乳腺癌細胞(Mda-157)時,也可以看到類似的事件(圖S1)。在所有的細胞模型系統中,最終的膜破壞導致細胞內Ca2+和fluo - 4am染料的損失,盡管動力學和擴展取決于所使用的鐵下垂誘導物和所研究的細胞系(圖1和S1)。胞漿Ca2+、細胞圓化和質膜破裂的增加均被鐵死亡抑制劑鐵他汀-1 (Fer- 1)特異性抑制,但壞死抑制劑壞死性他汀-1 (necc -1s)和泛半胱天酶抑制劑zVAD均未被抑制,表明所有這些事件都是鐵死亡的特征(圖1A F)。
與NIH-3T3細胞類似,fer1也完全抑制了RSL3處理后HT-1080和Mda-157細胞胞漿Ca2+的增加和細胞死亡(圖S1)。
我們通過流式細胞術平行追蹤fluo - 4am和PI熒光的時間過程(圖1G, H)。獨立于治療,胞漿Ca2+在質膜破裂前數小時就出現了增加。
在erastin -1處理的細胞中,我們發現fer1并沒有完全消除胞漿內Ca2+的增加(圖1A),約40%的細胞群保留fluo - 4am陽性(圖1C, G)。
相反,當RSL3誘導鐵死亡時,fer1完全阻止胞質Ca2+的增加(圖1B, D, H)。這些結果表明,Erastin誘導了兩種不同的Ca2+信號通路:一個與鐵死亡無關,另一個與鐵死亡特異。
胞質Ca2+的晚期增加可以被認為是鐵死亡的標志,因為它被fer1抑制,在Erastin-1或RSL3-誘導的鐵死亡中很常見。
二、脂質氧化先于胞質Ca2+的增加和質膜的破裂
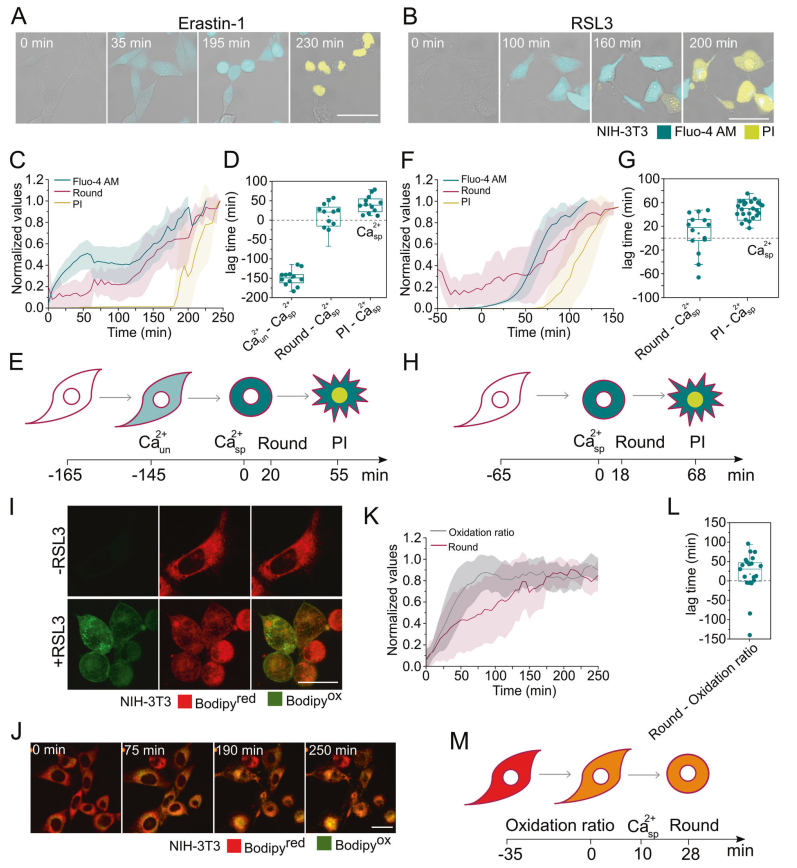
使用活細胞共聚焦顯微鏡在單細胞水平上量化了fluo - 4am信號的增加、細胞圓度和PI攝入。從單細胞動力學曲線(圖2C, F, K)來看,計算了實現每種鐵死亡表型50%變化(t50)所需的時間。
這使得我們可以設置鐵死亡過程中每個過程之間的延遲時間(圖2D, G, L)。
與使用的鐵晶體觸發器無關,胞質Ca2+的增加先于細胞圓化和完全的質膜塌陷(圖2A, B)。
然而,流式細胞術實驗表明(圖1G),在存在Erastin-1的情況下,細胞內Ca2+的增加是一個兩階段的過程,具有兩相行為(圖2C)。
第一個事件在最大Ca2+信號的40%左右達到飽和,對應于不相關的鈣(Ca2+ un)增加,因為它沒有被fer1抑制(圖1)。
第二種Ca2+升高,這是RSL3處理的常見現象,并被fer1抑制(圖1H和2F),對應于胞漿內Ca2+升高,特異性于鐵下垂(Ca2+ sp)。
在時間上,Ca2+ un在Erastin-1處理后增加,隨后是Ca2+ sp上升,細胞圓角和最終的PI攝入量(圖2C E)。
相比之下,rsl3處理的細胞具有獨特的特異性Ca2+升高事件,隨后是細胞圓整和最終的PI攝入(圖2F-H)。
為了觀察膜中的脂質過氧化,我們使用了脂質過氧化傳感器C11 BODIPY 581/591。RSL3處理確實促進了質膜和亞細胞膜的脂質過氧化,可以通過增加BODIPY (BODIPYox)的綠色熒光信號來檢測(圖2I)。接下來,我們平行追蹤氧化率和細胞圓角的變化(圖2J, K)。脂質過氧化先于細胞圓角。考慮到胞漿Ca2+的增加到細胞圓角的滯后時間始終低于脂質過氧化到細胞圓角的滯后時間,我們可以估計,Ca2+的增加發生在脂質過氧化之后(圖2M)。
這一估計是基于fluo - 4am增加的獨立動力學(圖2fh)和BODIPY氧化率(圖2K, L)之間的外推。
三、滲透活性藥物保護細胞免受鐵死亡
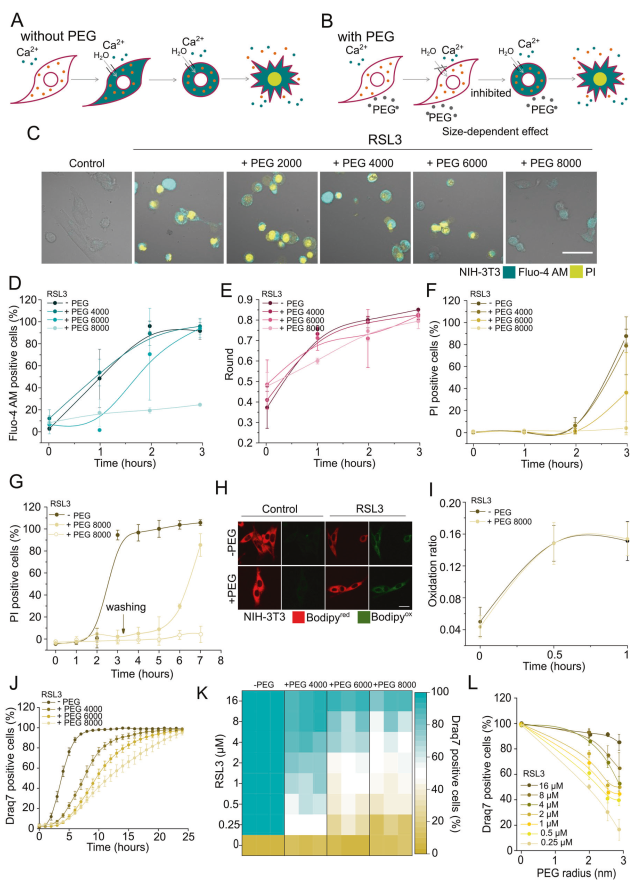
孔的形成是不同類型調控細胞死亡的共同特征,包括凋亡[29]、壞死[13]和焦亡[30]。質膜孔的打開導致水分子的凈流入,這是由于高濃度的不能通過膜孔的細胞內大分子造成的滲透不平衡的結果(圖3A)。可以通過添加適當大小的不能通過孔進入細胞的滲透保護劑來阻止這種影響,從而平衡細胞內滲透壓、水流入和隨之而來的細胞崩潰(圖3B)。由此可見,聚乙二醇(peg)的滲透作用可以調節膜孔的滲透性,而離子通道的滲透性則不受調節。為了研究在鐵死亡細胞中觀察到的質膜通透性是否與膜孔有關,我們評估了不同大小的peg對不同鐵死亡的動力學和程度的影響。
更小的peg的添加達到4000,并沒有阻止胞漿Ca2+的增加,細胞圓角或細胞死亡(圖3C F和S2)。
相比之下,高分子量peg(6000和8000)存在時,細胞漿Ca2+水平的升高和細胞死亡都以大小依賴的方式延遲(圖3D, F和S2D)。
在PEG 8000的存在下,我們還觀察到細胞舍圓動力學的延遲(圖3E)。這些結果表明,胞漿Ca2+的增加、細胞圓潤和質膜破裂都是由滲透力驅動的事件。值得注意的是,PEG 8000對鐵死亡的保護作用在洗滌后恢復,這表明隨著時間的推移,膜損傷是穩定的(圖3G)。我們還發現,PEG 8000不能防止rsl3處理的NIH-3T3細胞的脂質過氧化(圖3H, I)。
值得注意的是,對于所有使用的PEG大小,當NIH-3T3細胞處理較長時間(24小時)時,細胞死亡恢復(圖3J)。
peg對鐵死亡動力學的抑制作用也隨著RSL3濃度的增加而降低(圖3K、L和S3A)。
以在低濃度RSL3下能夠起到保護作用的PEG大小作為參考,我們可以粗略估計在2.5 nm半徑附近的質膜穿孔部分較小的尺寸(圖3L)。
四、ESCRT-III復合物在鐵死亡過程中被激活并拮抗細胞死亡
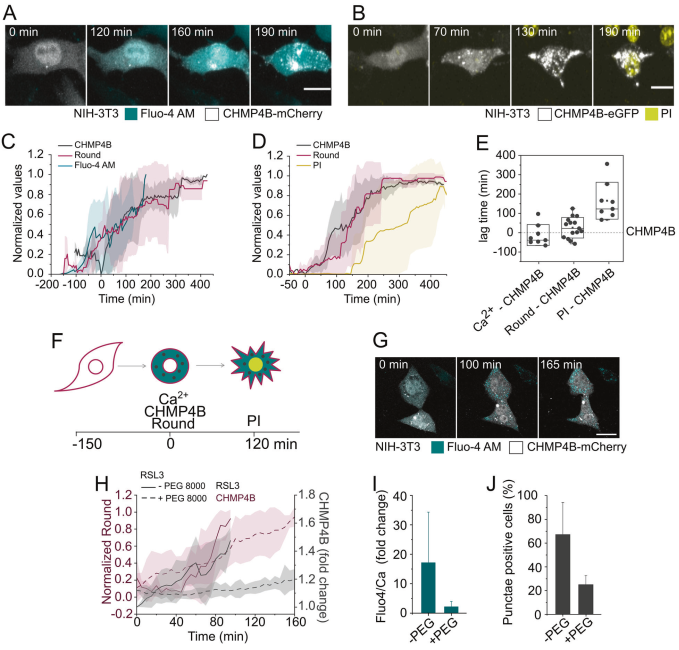
最初,CHMP4B主要表現為均勻的胞漿分布,然而,在RSL3處理后,蛋白定位于不同的斑點,隨著時間的推移,蛋白數量和熒光強度增加(圖4A、B和S4)。
這一觀察結果與報道的CHMP4B在壞死[22,23]和焦亡[17]中的行為相似,表明ESCRT-III復合體在鐵死亡過程中被激活。
接下來,我們通過活細胞共聚焦成像在單細胞水平上量化了CHMP4B斑點形成與細胞質Ca2+濃度增加、細胞圓角和最終質膜破裂的時間相關性。CHMP4B斑點的形成大致與胞質Ca2+濃度的增加相吻合(圖4C E和S4)。
此外,這些結果表明,細胞聚集在這兩個事件發生的同一時間(圖4C E),這與滲透力的假定作用是一致的。在細胞漿Ca2+水平持續升高、細胞圓化和esrt - iii復合體激活后發生最終的膜破裂,可能是當細胞修復機制最終被淹沒時(圖4B, E, F)。
PEG 8000也阻斷了CHMP4B斑點的形成,這可能是抑制Ca2+通量的結果(圖4G J)。
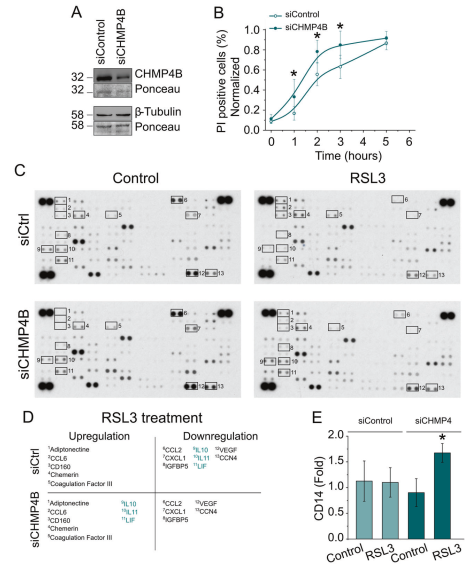
為了檢驗ESCRT-III在鐵死亡中的功能作用,我們評估了敲除CHMP4B對NIH-3T3細胞死亡的影響(圖5A, B)。
我們開始使用蛋白質組分析器XL細胞因子陣列試劑盒(圖5C和S6)來確定鐵下垂是否可以直接調節不同細胞系中的細胞因子分泌。
在鐵下垂的誘導下,我們檢測到出現細胞系和治療依賴的細胞因子亞群水平的變化。此外,敲除CHMP4B改變了RSL3(圖5C, D)或Erastin-1(圖S6C)處理后獲得的細胞因子譜。